A novel peptide microarray for protein detection and analysis utilizing a dry peptide array system
Kenji
Usui
a,
Kin-ya
Tomizaki
a,
Takafumi
Ohyama
b,
Kiyoshi
Nokihara
b and
Hisakazu
Mihara
*a
aDepartment of Bioengineering and the COE21 program, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, Nagatsuta-cho 4259 B-40, Midori-ku, Yokohama 226-8501, Japan. E-mail: hmihara@bio.titech.ac.jp; Fax: +81-45-924-5833; Tel: +81-45-924-5756
bHiPep Laboratories, Nakatsukasacho 486-46, Kamigyo-ku, Kyoto 602-8158, Japan
First published on 23rd December 2005
Abstract
A novel dry peptide microarray system has been constructed that affords a practical solution for protein detection and analysis. This system is an array preparation and assay procedure under dry conditions that uses designed peptides as non-immobilized capture agents for the detection of proteins. The system has several advantages that include its portability and ease-of-use, as well as the fact that vaporization of sample solutions need not be considered. In this study, various proteins have been characterized with an α-helical peptide mini-library. When proteins were added to the peptide library array, the fluorescent peptides showed different fluorescent intensities depending on their sequences. The patterns of these responses could be regarded as ‘protein fingerprints’ (PFPs), which are sufficient to establish the identities of the target proteins. Furthermore, statistical analysis of the resulting PFPs was performed using cluster analysis. The PFPs of the proteins were clustered successfully depending on their families and binding properties. Additionally, the target protein was characterized using a nanolitre system and could be detected down to 1.2 fmol. These studies imply that the dry peptide array system is a promising tool for detecting and analyzing target proteins. The dry peptide array will play a role in development of high-throughput protein-detecting nano/micro arrays for proteomics and ligand screening studies.
Introduction
Since the completion of genome sequencing of many organisms, vast amounts of genomic information for understanding cellular events have been acquired. In the post-genome era, knowledge based on genomics will be applied to numerous fields including proteomics, which will be one of the most important fields in biomedical and biotechnological research. One of the most powerful analytical tools to address problems in these fields is DNA microarray technology, which can analyze mRNA transcript levels expressed under various conditions.1,2 The mRNA expression levels, however, do not always correlate with the corresponding protein abundances.3 Furthermore, the analysis of mRNA expression, as well as DNA sequence, does not provide information about protein functions, their interactions, activities, 3D structures and post-translational modifications such as proteolysis, phosphorylation, glycosylation, alkylation, and so on. Therefore, proteins need to be analyzed directly in order to obtain such information. The present technologies such as 2D gel electrophoresis, chromatographic separation, surface plasmon resonance, and mass spectrometry methods,4–7 however, still lack the parallelized and miniaturized capabilities for high-throughput screening of proteins.A protein-detecting chip is expected to be one of these high-throughput and robust technologies for direct analyses of protein functions and interactions, and has become one of the significant research areas in biotechnology.8–19 In comparison to DNA or RNA, the complex structures and interactions of proteins are recognized as significant problems in achieving protein-detection chips. The selection of capture agents in array formats is one of the most critical points to approach these problems in an early phase of the construction of protein-detection chip technologies.
Designed peptide libraries with suitable secondary and/or tertiary structures are promising candidates for protein capturing agents, because of the following advantages: (a) peptides are more easily designed and synthesized than antibodies or recombinant proteins; (b) peptides with suitable structures can mimic protein–protein interactions and can be candidates for substrates and ligands;20–22 (c) probes for protein-detection such as fluorophores and linker groups for immobilization can be introduced into peptides at any required position; (d) not only naturally occurring amino acids but also various functional moieties such as artificial amino acids, sugars, alkyl groups and co-factors can be employed as building blocks in the designed peptides. So far, we have recently constructed designed peptide libraries with loop, α-helical or β-sheet structures in an attempt to satisfy these criteria.23–26 When various proteins were added to the arrays with these peptide libraries, the peptide spots showed different fluorescence responses for the proteins depending on their sequences. The patterns of these responses could be regarded as ‘protein fingerprints’ (PFPs), which are able to establish the identity of target proteins. The resulting PFPs correlated with the recognition properties of the proteins. Thus, it has been demonstrated that the designed peptide microarray is one of the most useful approaches for protein-chip technology.
In order to approach a more practical array format, the following two points are considered to be critical; the selection of wet or dry conditions for the array and the choice of immobilization or non-immobilization methods for the arraying capture agents. In the case of the former selection, denaturation of the capture agents (proteins) is usually a problem and hence preparation of a protein array is generally difficult in the conventional dry manner that has proved useful in studies with DNA chips. Therefore, wet or semi-wet conditions are regarded as an appropriate method for a protein array and some excellent work has been reported.11,19 The wet system has, however, still not been developed owing to the difficulty of manufacturing arrays. Using capture agents, other than proteins, under dry condition is one of the solutions for this problem. In the case of the latter, immobilization of capturing agents in an array preparation has still some problems such as cumbersome procedures and the nonexistence of an established method compared to DNA arrays, although recent studies using immobilization methods have been well represented.10,11,13 Hence, establishment of a novel non-immobilization method with a dry array format is meaningful and important.
The dry peptide array system is a novel system, which contains an array preparation and assay procedures with dried samples including non-immobilized capturing agents (designed peptides) (Fig. 1). The system has some advantageous characteristics as follows: (1) Designed peptides are useful candidates as capture agents for protein-detection as described above. (2) Peptides are tolerant of dry conditions. (3) The array is easily portable. (4) The preparation and assay procedures are much easier than those of immobilization methods. (5) No plate washing processes are required. (6) Vaporizing of the sample solution need not be taken into account in nanolitre solution measurements. (7) This system can be applied to the characterizations of simple proteins or analyses of binding properties for proteomics studies and ligand screening applications with easy and robust fashion. Here, we describe the construction of a novel protein-detection system with an α-helical peptide mini-library using the dry peptide array system. Initially, in order to detect various proteins including the EF-hand family and interferons, a mini-library consisting of 20 fluorescent peptides with various charges and/or hydrophobicities was constructed. Then, the PFPs of proteins were obtained with the fluorescent responses of the library peptides using the dry peptide array system. Statistical data generated from PFPs were also analyzed using a cluster method. Moreover, the detection of target proteins using the dry peptide array method on the nanolitre scale with a robotic spotter was demonstrated. The present study shows that the dry peptide array system using the PFP method will allow realization of high-throughput protein nano/micro arrays for proteomic analyses and ligand screenings.
 | ||
| Fig. 1 (A) Illustration of the micro-well glass plates used in this study. (B) Scheme of the dry peptide array system with the micro-well plates. (C) Illustration of printing mechanism using a Micro Spotting Pin of SpotBot (CMP10, TeleChem International) for sample spotting. (D) Scheme of the dry peptide array system at the nanolitre level. | ||
Results and discussion
Design and synthesis of an α-helical peptide mini-library for the dry peptide array system
A mini-library consisting of 20 designed α-helical peptides with various charges and/or hydrophobicities was constructed to detect a variety of proteins including calmodulin (CaM). The strategy for the construction of the α-helical peptide mini-library is shown in Fig. 2. On the basis of the L8K6 sequence, which is a basic amphiphilic α-helical peptide with the sequence LKKLLKLLKKLLKL27 and which is known to bind to CaM in the presence of Ca2+,28,29 the eight Leu residues were replaced systematically with Ala, and the six Lys residues were also replaced systematically with Glu. This mini-library was a subset of the α-helical peptide library described previously,24,25 and the library was synthesized as described in the Experimental section. | ||
| Fig. 2 (A) Strategy for the construction of the α-helical peptide mini-library. The sequence of No. 1 L8K6 peptide and construction of α-helical peptide mini-library (upper side), diagrams of α-helix structure with a wheel representation of L8K6 (lower-left side), the structure of TAMRA (lower-right side). (B) Numbers and names of peptides in the α-helical peptide mini-library. | ||
Structural analysis using far-UV CD spectroscopy and FT-IR spectroscopy
In order to estimate the structural features of the peptides, the far-UV CD spectra of designed peptides were measured, as shown in Fig. 3. Representative peptides (No. 1, 4, 8, 9, and 20) displayed negative minima with ellipticities at 208 nm and 222 nm, which are characteristic of an α-helical structure.30 FT-IR spectra were also measured to estimate the structural details of the peptides under the dry conditions. Representative peptides (No. 1 and 9) displayed absorption maxima at amide I (1654 cm−1) and amide II (1546 cm−1), which are characteristics of an α-helical structure.31,32 These results indicated that the peptides themselves assumed an α-helical structure under the dry conditions as well as in the aqueous solution, although a variety of α-helical propensities were observed depending on the sequences.![Circular dichroism spectra of representative designed α-helical peptides (No. 1, 4, 8, 9 and 20). [Peptide] = 5.0 µM in 20% TFE, 20 mM Tris–HCl, 0.5 mM CaCl2, pH 7.4 at 25 °C.](/image/article/2006/MB/b514263f/b514263f-f3.gif) | ||
| Fig. 3 Circular dichroism spectra of representative designed α-helical peptides (No. 1, 4, 8, 9 and 20). [Peptide] = 5.0 µM in 20% TFE, 20 mM Tris–HCl, 0.5 mM CaCl2, pH 7.4 at 25 °C. | ||
Detection of CaM using representative peptides in solution (solution validation)
To confirm whether TAMRA-labeled peptides were suitable for detection of CaM, the fluorescence spectra of representative peptides (No. 1, 4, 8, 9, and 20) and the changes in fluorescence upon addition of CaM were measured in the buffer solution (Fig. 4). From the increase in fluorescence (ca. 3 fold on addition of 8.0 µM CaM), which might be ascribable to the conformational changes of α-helix peptides upon binding to proteins, the binding constant of No. 1 L8K6 peptide with CaM was calculated as 0.93 × 106 M−1 by means of a single site binding equation33 (Fig. 4A). The binding constants of No. 4 and 9 could be also calculated as 0.12 × 106 M−1 and 0.64 × 106 M−1, respectively, while the binding constants of No. 8 and 20 could not be estimated because of their weak binding (Fig. 4B). The results are related to the number of Lys and Leu residues and consistent with those of previous work in solution.24 The data show that No. 1 and 9 peptides have higher affinity than No. 4, 8 and 20 which is compatible with the known CaM binding propensities i.e. CaM preferably binds to a highly cationic α-helix peptide with hydrophobic Leu residues.27,28 | ||
| Fig. 4 (A) A fluorescence titration curve of No. 1 L8K6 (1.0 µM) upon addition of CaM in 20 mM Tris–HCl, 0.1 mM CaCl2, 150 mM NaCl, 20 mM PEG2000, pH 7.4 at 25 °C. λex 554 nm, λem 585 nm. (B) Binding constants of the representative peptides (No. 1, 4, 8, 9, and 20) determined by fluorescence changes of the peptides upon addition of CaM in solution. N.D. denotes that the Ka value is not determined precisely as fluorescence intensity changes are small. | ||
Characterization of CaM utilizing the dry peptide array system (dry array validation)
CaM was further characterized with the α-helical peptide library utilizing the dry peptide array system. The procedure (Fig. 1B) was as follows. The peptide solution was spotted into the wells of the glass plates and the spotted solution was dried using a vacuum pump. Subsequently, the protein solution or a reference solution was spotted and dried peptides were dissolved. After 15 min in a humidistat at room temperature (time for redissolving peptides and binding them to proteins), the solution was dried using a pump. The responses to the addition of the protein were evaluated using the values (I/I0) calculated as the fluorescence intensity with protein (I) against that without protein (I0). These fluorescence increases might be ascribable to the conformational changes of α-helix peptides upon binding to proteins. The peptides showed sequence dependent responses upon addition of CaM (75 ng, 4.5 pmol) (Fig. 5A). Some peptides (No. 6, 9, 13, etc.) showed higher fluorescence responses (3–4 fold), and some (No. 8, 16, 20, etc.) gave responses of less than 2 fold. To understand the binding properties of CaM more easily, the pattern of these fluorescence responses was converted into a gray image, regarded as a ‘protein fingerprint’ (PFP) as shown in Fig. 5B. The gray color of each cell displays the response of each peptide towards CaM. The cells in peptide No. 9 and its vicinity were white and light gray. According to these results, CaM prefers to bind cationic amphiphilic peptides with abundant Leu residues. It is correlated with the results of previous work in solution24 the same as in the previous section (Fig 4B). A protein fingerprint of apoCaM (CaM (75 ng, 4.5 pmol) with 150 pmol EDTA) was also obtained (Fig. 5C). This PFP was very different to that of CaM (75 ng, 4.5 pmol) with free Ca2+. This indicated that in the absence of Ca2+, apoCaM can bind different peptides due to a structural change of CaM itself, and that the fingerprint depends on the protein tertiary structure.![(A) A fluorescence response pattern of the α-helical peptide mini-library upon addition of 75 ng CaM with the dry peptide array system. λex 543 nm, λem 570 nm. [Peptide] = 1.5 pmol. (B) The ‘protein fingerprint’ (PFP) of CaM (75 ng, 4.5 pmol). (C) The PFP of apoCaM (CaM (75 ng, 4.5 pmol) with 150 pmol EDTA). (D) The PFPs of CaM (750 ng, 7.5 ng). (E) A scanned image of L8K6 (No. 1) and L6A2K4E2 (No. 9) upon addition of CaM at various concentrations. (F) Fluorescence intensities of L8K6 (No. 1) and L6A2K4E2 (No. 9) upon addition of CaM at various concentrations. All data show the average of 5 measurements.](/image/article/2006/MB/b514263f/b514263f-f5.gif) | ||
| Fig. 5 (A) A fluorescence response pattern of the α-helical peptide mini-library upon addition of 75 ng CaM with the dry peptide array system. λex 543 nm, λem 570 nm. [Peptide] = 1.5 pmol. (B) The ‘protein fingerprint’ (PFP) of CaM (75 ng, 4.5 pmol). (C) The PFP of apoCaM (CaM (75 ng, 4.5 pmol) with 150 pmol EDTA). (D) The PFPs of CaM (750 ng, 7.5 ng). (E) A scanned image of L8K6 (No. 1) and L6A2K4E2 (No. 9) upon addition of CaM at various concentrations. (F) Fluorescence intensities of L8K6 (No. 1) and L6A2K4E2 (No. 9) upon addition of CaM at various concentrations. All data show the average of 5 measurements. | ||
Furthermore CaM at various concentrations showed similar responses although small differences in the patterns were present (Fig. 5D), and the fluorescence intensities linearly increased with the increase of CaM concentration up to 30 pmol. The fluorescence changes of representative peptides (No. 1 and 9) in the library are shown (Fig. 5E and 5F), indicating that the fluorescence intensities for CaM are dose-dependent with less than 7.5 ng (450 fmol) as the detection limit and the responses were highly reproducible. These results implied that the concentration of CaM could be evaluated from both the protein fingerprints and the fluorescence intensities.
Characterization of various proteins utilizing the dry peptide array system
A variety of proteins could also be characterized with the α-helical peptide library utilizing the dry peptide array system. Representative protein samples were selected as follows: CaM, S-100 proteins (S-100), myosin as model proteins containing EF-hand motifs, human interferon-α (huIFN-α), human interferon-γ (huIFN-γ), rat interferon-γ (ratIFN-γ) as model proteins of cytokine (purchased from TOYOBO), insulin as a model of small proteins, and α-amylase as a model of glycosidase. The responses upon addition of the proteins were evaluated using the values (I/I0). Gray scale image ‘protein fingerprints’ (PFPs) are shown in Fig. 6. It is obvious that the PFPs of proteins with EF-hand motifs are quite different amongst themselves and different from the IFN family. Clearly PFPs are characteristic and are able to identify each protein uniquely.![PFPs of various proteins (75 ng). [Peptide] = 1.5 pmol. S-100: S-100 proteins, huIFN-α: human interferon-α, huIFN-γ: human interferon-γ, ratIFN-γ: rat interferon-γ. All data show the averages of 5 measurements.](/image/article/2006/MB/b514263f/b514263f-f6.gif) | ||
| Fig. 6 PFPs of various proteins (75 ng). [Peptide] = 1.5 pmol. S-100: S-100 proteins, huIFN-α: human interferon-α, huIFN-γ: human interferon-γ, ratIFN-γ: rat interferon-γ. All data show the averages of 5 measurements. | ||
As far as the library peptides were concerned, the No. 9 peptide, for example, seemed to be one of the best lead sequences of binder to CaM, S-100, or myosin, although various changes of fluorescence intensity in No. 9 peptide were observed depending on proteins. A rigid structure might influence these binding results, because No. 9 peptide showed a high α-helical content in the CD and FT-IR measurements. The result implies that the dry array method can be applicable to ligand screening.
Data mining of PFPs
The similarities between the different PFPs were analyzed quantitatively by the method of the Euclidian distances.34,35 The distance matrix (Fig. 7A) was generated by the statistical treatment of normalized PFPs (Fig. 7C). The values for the similarity distances between the normalized PFP patterns were determined by Euclidian distance in multidimensional space defined by each PFP data and represented by gray scale (white for the highest similarity and black for the lowest). For example, the similarity distance between CaM and S-100 (CaM/S-100) was 11.9, showing a light gray block in the distance matrix. On the other hand, the distances of CaM/apoCaM and CaM/huIFN-α were 24.8 and 33.8, respectively, showing a dark gray or a black block on the matrix. Thus, the PFP patterns of CaM/S-100 were evaluated as similar, while the PFPs of CaM/apoCaM and CaM/huIFN-α were very different from each other. It can be seen that the PFPs of EF-hand proteins have a similarity but are significantly different from those of other protein families and that the PFP depends on the protein secondary/tertiary structure.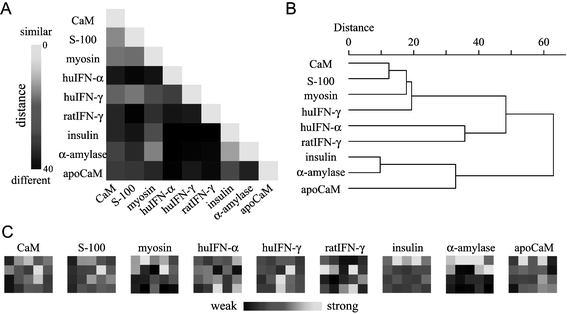 | ||
| Fig. 7 (A) The Euclidian distance matrix between the various PFPs. (B) The dendrogram of various PFPs generated by the analysis of the Euclidian distances. (C) Normalized PFPs of various proteins. | ||
Furthermore, a cluster analysis among the PFPs was conducted. The clustering dendrogram (Fig. 7B) was generated by the analysis of Euclidian distances. The horizontal axis indicates the distances among PFPs (left for PFPs with the highest similarity and right for PFPs with the lowest similarity). The analyses showed that proteins within an EF-hand family are clustered successfully. On the other hand, apoCaM was distinguished from EF-hand family proteins including CaM (with Ca2+). This indicated that the structural change could be monitored by PFPs. Additionally, this system could distinguish not only huIFN-α from huIFN-γ but also human IFN-γ from rat IFN-γ, while the IFNs were clustered within a relatively narrow range. With an improvement in the peptide library, such as using a variety of amino acid residues or applying peptides with other structures, these proteins could be clustered more precisely depending on their structures or subtypes. These results implied that PFPs using designed peptides with a suitable secondary structure might characterize the nature and function of a variety of proteins.
The nano detection of CaM with the dry peptide array
The detection of CaM on a nanolitre scale (3.9 nl spot volume, 8 fmol peptide per spot) with a robotic spotter dispensing representative peptides (No. 1, 4 and 8) was performed. The procedure is described in the Experimental section and shown in Fig. 1C and 1D. With the nano dry peptide array system, CaM was also characterized by the fluorescence changes of peptides (Fig. 8A) as in the micro detection described above, indicating that the fluorescence intensities for CaM are dose-dependent with 20 pg (1.2 fmol) as the detection limit and the responses were highly reproducible. Furthermore, adding EDTA prevented the responses (Fig. 8B). This result was coincident with that in solution, apoCaM being discriminated from native CaM. The responses to insulin were also obtained and its pattern was very different from that of CaM (the fluorescent intensities of insulin were lower than those of CaM) (Fig. 8C). These results imply that the nanolitre-size dry peptide array system and resulting PFPs are also available for protein-detection and are similar to the microlitre and solution systems. | ||
| Fig. 8 (A) A scanned image and fluorescence intensities of L8K6 (No. 1) upon addition of CaM at various concentrations. (B) Fluorescence intensities of L8K6 (No. 1) upon addition of CaM (200 pg) with or without 400 fmol EDTA. (C) Fluorescence intensities of No. 1, 4 and 8 upon addition of CaM (200 pg) or insulin (200 pg). | ||
Conclusions
In the present study, we have demonstrated the successful construction of a dry peptide array system in a format suitable for use as a novel protein detection chip. This system is an array preparation and assay procedure with dried samples involving non-immobilized designed peptides (capture agents). The results of the dry array appeared to be correlated with those of the solution assay and the detection limit was less than 20 pg (1.2 fmol) of target protein with the nanolitre system. The dry peptide array system is easy to handle, very portable and affords reproducible results. Additionally, any plate washing processes are not required, because the bindings of the library peptides to the target proteins can be observed with changes of fluorescence intensities of the peptides. These features imply that the technology could be a robust system for arraying capture agents as an alternative to the immobilization method, which has recently been applied successfully in biochip studies. Moreover, data treatment which is indispensable for analyzing various properties of target proteins in protein chip technology was successfully demonstrated. We have conducted statistical data treatments using Euclidian distance and cluster analyses, which are measures of the similarity among protein fingerprint (PFP) patterns. Using a small library of several dozen peptides, proteins in the same family could be uniquely characterized from their PFPs. 3D structural changes within a protein could also be monitored. This suggests that the peptide microarrays with PFPs will be a robust analytical tool for probing the binding and structural properties of proteins. Thus this study shows that the dry peptide array system with designed peptides using the PFP method has great promise for the realization of a high-throughput protein nano/micro array system for proteomics and ligand screening applications.Experimental
General remarks
The micro-well glass plates were provided by Nippon Sheet Glass Co., Ltd. Arrays consisting of 50 holes of 2.5 mm in diameter were etched on the glass plate (25 mm × 75 mm × 1.05 mm) (Fig. 1A). All chemicals and solvents were of reagent or HPLC grade and were used without further purification. Protein samples were purchased from Sigma-Aldrich Japan unless otherwise stated. HPLC was performed on a Hitachi L7000 or a Shimadzu LC2010C system using a YMC-Pack ODS-A (4.6 × 150 mm) for analysis, and a YMC ODS A323 (10 × 250 mm) for preparative purification with a linear gradient of acetonitrile/0.1% trifluoroacetic acid (TFA) at a flow rate of 1.0 ml min−1 for analyses and 3.0 ml min−1 for preparative separation, respectively. MALDI-TOF MS was measured on a Shimadzu KOMPACT MALDI III with 3,5-dimethoxy-4-hydroxycinnamic acid as the matrix. Amino acid analysis was carried out using a Wakopak WS-PTC column (4.0 × 200 mm; Wako Pure Chemical Industries) after hydrolysis in 6 M HCl at 110 °C for 24 h in a sealed tube followed by phenyl isothiocyanate labeling.Synthesis of an α-helix peptide mini-library
The designed peptide library was synthesized on Rink amide resin by combination of automatic (Advanced ChemTech Model 348 MPS) and manual syntheses with Fmoc chemistry.36 Side chain protections were as follows: t-butyl (tBu) for Glu; t-butyloxycarbonyl (Boc) or 4-methyltrityl (Mtt) for Lys. Initially, Fmoc–Gly-resin was synthesized with Fmoc–Gly–OH (1/3 eq.) by the manual method using 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 1/3 eq.), 1-hydroxybenzotriazole (HOBt, 1/3 eq.) and diisopropylethylamine (DIEA, 2/3 eq.) to reduce the loading amount of resin (0.25 mmol g−1). Then, Ac–Lys(Mtt)-[various sequences]-Gly-resins were synthesized using the synthesizer with Fmoc–AA–OH (10 eq.) by the HBTU–HOBt method. After assembly of all peptides, the Mtt group was removed by treatment with dichloromethane–triisopropylsilane–trifluoroacetic acid (TFA) (94 ∶ 5 ∶ 1, v/v) and the 5-(and-6)-carboxytetramethylrhodamine (TAMRA) moiety was introduced by using TAMRA (1.5 eq.) with O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU, 1.5 eq.) and diisopropylethylamine (DIEA, 3 eq.). The peptides with fluorescent probes were cleaved from the resin and side chain protections were removed with TFA–H2O (20 ∶ 1, v/v) for 2 h. The peptides were precipitated by addition of diethylether and collected by centrifugation. The crude peptides were purified by RP-HPLC and characterized by MALDI-TOF MS and amino acid analysis. The purified peptides were dissolved in methanol at ca. 1 mM, and stored at 4 °C. In order to estimate the concentration of the stock solution of the library peptides, peptide absorbance was measured on a Benchmark Multiplate Reader (Bio-Rad Laboratories) with a 490 nm filter using microtiter plates (Assay Plate, IWAKI) in 50 mM KH2PO4 containing 5.0 M guanidine hydrochloride (pH 9.0), and compared with that of a standard peptide, L8K6, whose concentration was determined by amino acid analysis.Spectroscopic measurements
The fluorescence measurements were performed in 20 mM Tris–HCl containing 0.1 mM CaCl2, 150 mM NaCl, 20 mM PEG2000 (pH 7.4). Fluorescence spectra were recorded on a Hitachi F-2500 fluorescence spectrophotometer with a thermoregulator using a quartz cell with 10 mm pathlength at 25 °C. Fluorescence intensities of the fluorescently-labeled peptides in micro-well plates were measured by a Scan Array 4000 XL (GSI Lumonics) at room temperature. 543 nm and 570 nm filters were used for excitation and emission, respectively. Fluorescence intensities of the fluorescently-labeled peptides in nano-spotted plates were measured by a CRBIO IIe (Hitachi Software Engineering) at room temperature. 532 nm and 573 nm filters were used for excitation and emission, respectively. Circular dichroism (CD) measurements were performed in 20% TFE, 20 mM Tris–HCl containing 0.5 mM CaCl2 (pH 7.4) on a Jasco J-720WI spectropolarimeter with a thermoregulator using a quartz cell with 1 mm pathlength at 25 °C. Fourier transform infrared (FT-IR) spectroscopy was carried out on an IR Prestige-21 (Shimadzu). The peptide solution in MeOH (25 µl, ca. 0.2 mM) was placed on a CaF2 plate (ϕ 13 mm) and dried using a vacuum pump (ca. 10 Pa, 15 min) at room temperature. This was performed twice and then H2O (25 µl) was dropped onto the peptide film and dried using a pump (ca. 10 Pa, 2 h) at room temperature.Protein detection assay using the dry peptide array
The outline of the procedure is shown in Fig. 1B. The peptide solution (1.5 µl, 1.0 µM in H2O) was spotted into the wells of the glass plates manually and the spotted solution was dried using a vacuum pump (ca. 10 Pa, 15 min). Subsequently, the protein solution (1.5 µl, various concentrations of proteins with 100 µM CaCl2 in H2O) or a reference solution (1.5 µl, 100 µM CaCl2 in H2O) was spotted on each well, where peptides had been spotted and dried up, and dried peptides were dissolved. After 15 min in a humidistat at room temperature (time for redissolving peptides and binding them to proteins), the solution was dried using a pump (ca. 10 Pa, 15 min). The fluorescence intensities of the spots containing peptide alone (I0) and the spots containing peptide and protein (I) were measured. The peptide response to the proteins was expressed using the intensity ratio, I/I0. All data shown are the averages of 5 measurements.Data treatment for gray scale ‘protein fingerprints’
Data for ‘protein fingerprints’ (PFPs) in color (black–red–yellow) were manipulated according to the standard procedure reported previously.23–26,37 In this paper, the obtained PFPs were converted into PFPs in gray scale. The file format used here was portable-pixel-map (.ppm) format. Each grid position was first assigned as three whole numbers corresponding to RGB color-codes representing increment response (0, 0, 0) (full black, minimum increasing value) to (255, 0, 0) (red) to (255, 255, 0) (yellow, maximum increasing value), which corresponds to all the fluorescence change rates (I/I0) divided into 511 levels. The numbers of the grid were saved as a comma-separated-value (.csv) file including the three (or four) lines of ppm setting at the top of the file. The file was then saved in the portable-pixel-map format by simply adding ‘.ppm’ to the filename. This file was opened by a graphic viewer software, resized and saved in other formats such as bitmap file format (.bmp). Then the color-scale in the file was converted into gray scale (white for the highest similarity and black for the lowest).Data treatment using statistical analyses
The measure that we have used to determine the similarity between two PFPs obtained from different target proteins is the Euclidian distance.26,34,35 This is a common measure when considering the distance between two vectors. Before the Euclidian distance analyses were performed, the PFP had to be normalized. The normalization formula was as follows:| (Pori − Pave)/d = Pnorm |
The similarity between the normalized PFP patterns is measured by the Euclidian distance in multidimensional space defined by each PFP and represented by gray scale (white for the highest similarity and black for the lowest) converted from color coding (yellow for the highest similarity and black for the lowest) as described in the previous section. Additionally, a cluster analysis among the normalized PFPs was conducted. We have used Ward's clustering algorithm and the dendrogram was obtained with the analyses of Euclidian distances using the Excel Macro program.38 The horizontal axis represents the distance among normalized PFPs (left for PFPs with the highest similarity and right for PFPs with the lowest similarity).
The dry peptide array using a nano spotter
The outline of the procedure is shown in Fig. 1C and 1D. The peptide solution (3.9 nl, 2.0 µM peptide in H2O) was spotted onto flat glass plates, SuperAmine Microarray substrates (TeleChem International) using a SpotBot Personal Microarrayer (TeleChem International) (the spotted solution was dried spontaneously for a few minutes). Then, the protein solution (3.9 nl, various concentrations of protein with 100 µM CaCl2 in H2O) and the reference solution (3.9 nl, 100 µM CaCl2 in H2O) was arrayed on the spots of the peptides (the spotted solution also dried spontaneously). The fluorescence intensities of the spots containing peptide alone (I0) and the spots containing peptide and protein (I) were measured. The peptide response to the proteins was expressed using the intensity ratio, I/I0.Acknowledgements
The authors thank Dr Eiry Kobatake, Department of Biological Information, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, Japan, for measurements of the array scanner, and Dr Victor Wray, German Research Centre for Biotechnology, Braunschweig, for his linguistic assistance and discussions. The micro-well glass plates were generously provided by Nippon Sheet Glass Co., Ltd. This study was in part supported by grants of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and Nippon Sheet Glass Foundation. K. U. is grateful for Research Fellowships of the Japan Society for the Promotion of Science (JSPS) for Young Scientists.References
- C. M. Niemeyer and D. Blohm, Angew. Chem., Int. Ed., 1999, 38, 2865–2869 ( Angew. Chem. , 1999 , 111 , 3039–3043 ) CrossRef CAS.
- J. L. DeRisi, V. R. Iyer and P. O. Brown, Science, 1997, 278, 680–686 CrossRef CAS.
- S. P. Gygi, Y. Rochon, B. R. Franza and R. Aebersold, Mol. Cell. Biol., 1999, 19, 1720–1730 CAS.
- N. L. Anderson and N. G. Anderson, Electrophoresis, 1998, 19, 1853–1861 CrossRef CAS.
- A. Shevchenko, O. N. Jensen, A. V. Podtelejnikov, F. Sagliocco, M. Wilm, O. Vorm, P. Mortensen, A. Shevchenko, H. Boucherie and M. Mann, Proc. Natl. Acad. Sci. USA, 1996, 93, 14440–14445 CrossRef CAS.
- S. P. Gygi, B. Rist, S. A. Gerber, F. Turecek, M. H. Gelb and R. Aebersold, Nat. Biotechnol., 1999, 17, 994–999 CrossRef CAS.
- T. Natsume, H. Nakayama and T. Isobe, Trends Biotechnol., 2001, 19(Suppl.), S28–S33 CrossRef CAS.
- K.-Y. Tomizaki, K. Usui and H. Mihara, ChemBioChem, 2005, 6, 782–799 CrossRef CAS.
- T. Kodadek, Chem. Biol., 2001, 8, 105–115 CrossRef CAS.
- G. MacBeath and S. L. Schreiber, Science, 2000, 289, 1760–1763 CAS.
- P. Arenkov, A. Kukhtin, A. Gemmell, S. Voloshchuk, V. Chupeeva and A. Mirzabekov, Anal. Biochem., 2000, 278, 123–131 CrossRef CAS.
- H. Zhu and M. Snyder, Curr. Opin. Chem. Biol., 2001, 5, 40–45 CrossRef CAS.
- H. Zhu, M. Bilgin, R. Bangham, D. Hall, A. Casamayor, P. Bertone, N. Lan, R. Jansen, S. Bidlingmaier, T. Houfek, T. Mitchell, P. Miller, R. A. Dean, M. Gerstein and M. Snyder, Science, 2001, 293, 2101–2105 CrossRef CAS.
- M. F. Templin, D. Stoll, M. Schrenk, P. C. Traub, C. F. Vöhringer and T. O. Joos, Trends Biotechnol., 2002, 20, 160–166 CrossRef CAS.
- P. Mitchell, Nat. Biotechnol., 2002, 20, 225–229 CrossRef CAS.
- D. S. Wilson and S. Nock, Angew. Chem., Int. Ed., 2003, 42, 494–500 ( Angew. Chem. , 2003 , 115 , 510–517 ) CrossRef CAS.
- D. Kambhampati, Protein Microarray Technology, Wiley-VCH, Weinheim, 2003 Search PubMed.
- E. T. Fung, Protein Arrays: Methods and Protocols; Methods in Molecular Biology, Humana Press, New Jersey, 2004, vol. 264 Search PubMed.
- S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58–64 CrossRef CAS.
- Q. Zhu, M. Uttamchandani, D. Li, M. L. Lesaicherre and S. Q. Yao, Org. Lett., 2003, 5, 1257–1260 CrossRef CAS.
- C. Hultschig and R. Frank, Mol. Diversity, 2004, 8, 231–245 Search PubMed.
- S. Watanabe, K. Usui, K.-Y. Tomizaki, K. Kajikawa and H. Mihara, Mol. BioSyst., 2005, 1, 363–365 RSC.
- M. Takahashi, K. Nokihara and H. Mihara, Chem. Biol., 2003, 10, 53–60 CrossRef CAS.
- K. Usui, M. Takahashi, K. Nokihara and H. Mihara, Mol. Diversity, 2004, 8, 209–218 Search PubMed.
- K. Usui, T. Ojima, M. Takahashi, K. Nokihara and H. Mihara, Biopolymers, 2004, 76, 129–139 CrossRef CAS.
- K. Usui, T. Ojima, K.-Y. Tomizaki and H. Mihara, NanoBiotechnology, 2005, 1, 191–200 Search PubMed.
- J. A. Cox, M. Comte, J. E. Fitton and W. F. DeGrado, J. Biol. Chem., 1985, 260, 2527–2534 CAS.
- K. T. O'Neil and W. F. DeGrado, Trends Biochem. Sci., 1990, 15, 59–64 CrossRef CAS.
- Y. Maulet and J. A. Cox, Biochemistry, 1983, 22, 5680–5686 CrossRef CAS.
- N. Greenfield and G. D. Fasman, Biochemistry, 1969, 8, 4108–4116 CrossRef CAS.
- R. C. Cantor and R. S. Schimmel, Biophysical Chemistry, Part II: Techniques for the study of biological structure and function, W. H. Freeman and Company, New York, 1980 Search PubMed.
- W. K. Surewicz, H. H. Mantsch and D. Chapman, Biochemistry, 1993, 32, 389–394 CrossRef CAS.
- T. Kuwabara, A. Nakamura, A. Ueno and F. Toda, J. Phys. Chem., 1994, 98, 6297–6303 CrossRef.
- J. P. Goddard and J. L. Reymond, J. Am. Chem. Soc., 2004, 126, 11116–11117 CrossRef CAS.
- J. Grognux and J. L. Reymond, ChemBioChem, 2004, 5, 826–831 CrossRef CAS.
- W. C. Chan and P. D. White, Fmoc solid phase peptide synthesis: A practical approach, Oxford University Press, New York, 2000 Search PubMed.
- D. Wahler, F. Badalassi, P. Crotti and J. L. Reymond, Chem.–Eur. J., 2002, 8, 3211–3228 CrossRef CAS.
- http://aoki2.si.gunma-u.ac.jp/lecture/stats-by-excel/vba/html/clustan.html (Japanese).
| This journal is © The Royal Society of Chemistry 2006 |