Simple and versatile methods for the fabrication of arrays of live mammalian cells†
Thomas
Peterbauer
*a,
Johannes
Heitz
b,
Michael
Olbrich
b and
Steffen
Hering
a
aUniversity of Vienna, Department of Pharmacology and Toxicology, Althanstrasse 14, Vienna, 1090, Austria. E-mail: Thomas.Peterbauer@univie.ac.at; Fax: +43 1 4277 9553; Tel: +43 1 4277 55306
bJohannes-Kepler University Linz, Institute of Applied Physics, Altenbergstrasse 69, Linz, 4040, Austria
First published on 8th May 2006
Abstract
Single-step methods for the generation of patterned surfaces on hydrogels are presented. Poly(vinyl alcohol) films covalently bonded on glass cover slips and commercially available hydrogel-coated polystyrene plates were used as cell-repellent surfaces. Cell-adhesive domains were created by spotting dilute solutions of sodium hypochlorite onto the surfaces. Alternatively, domains supporting cell attachment were created by exposure to UV light from a xenon excimer lamp, employing a contact mask. Rat skeletal myoblast cells, HEK 293 human embryonic kidney cells and Caco-2 colon carcinoma cells adhered and spread exclusively on modified areas. The surfaces are durable for weeks under cell culture conditions and re-usable after removal of the cells by trypsin treatment. Arrays of adhesive spots seeded with cells at a low density permitted dynamic monitoring of cell proliferation. Selected colonies can be harvested from the surfaces by means of local trypsination. Thus, these techniques may provide useful tools for the isolation of clonal cell populations. Additionally, we demonstrate the possibility of surface-mediated gene delivery from the micro patterns. We show that DNA, complexed with a lipid reagent, can be adsorbed on modified poly(vinyl alcohol) coatings, resulting in spatially controlled adhesion and reverse transfection of HEK 293 cells.
Introduction
Engineering surfaces to create micro patterns of areas that either promote or impede attachment of mammalian cells is important for the study of factors that affect cell adhesion, shape and migration of cells.1 Cell–matrix interactions and changes in cell geometry in turn affect differentiation2 and even apoptosis.3 In addition, patterned surfaces are valuable tools for the miniaturisation of techniques, including the development of microfluidic platforms and cell-based arrays for high-throughput analysis of cellular functions.4,5Microcontact printing of cell-adhesive alkanethiols on gold surfaces is one of the most popular techniques to pattern cells.6 Regions between the cell-adhesive areas are filled with ethylene glycol-terminated alkanethiols to prevent attachment. Various alternative strategies have been proposed, including spatially controlled deposition of proteins, peptides or extracellular matrix components to create adhesive domains on cell-repellent surfaces4,7 or, vice versa, deposition of poly(ethylene glycol) or poly(ethylene oxide) based polymers to prevent adhesion on bioactive surfaces.8,9 Areas promoting cell attachment have also been created by ablation of anti-adhesive polymers10 or by functionalization of cell-repellent polymeric materials by plasma11 or UV light.12
In this report, we present simple and versatile strategies to create micro patterns on hydrogels. We utilize poly(vinyl alcohol) (PVA) coatings on glass and a commercially available hydrogel covalently bonded onto polystyrene as cell-repellent surfaces. These hydrophilic polymers effectively prevent attachment of anchorage-dependent mammalian cells because adsorption of cell adhesion proteins such as fibronectin and vitronectin is minimal.13 Cell-adhesive areas can be created by oxidation of the surfaces using dilute sodium hypochlorite. The described method is unique due to the simplicity of the process, which is inexpensive and can be realized without any specialized equipment. Alternatively, the repellent surfaces can be photopatterned using UV light, allowing the creation of patterns of adhesive areas in combination with photo masks. We demonstrate that seeding cells at a very low density on a high density array of adhesive spots permits monitoring of the proliferation of single cells. Since methods for harvesting of cells from small areas by local trypsination are available, this strategy may provide a useful tool for the isolation of clonal cell populations.
In addition, we show that the patterned surfaces are compatible with substrate-mediated gene delivery (“reverse transfection”). This technique, originally developed by Ziauddin and Sabatini,14 enables high-throughput analysis of gene function in live mammalian cells. It uses sets of cDNAs in expression vectors printed on defined areas of glass slides. Upon addition of transfection reagents and cells, clusters of cells expressing the gene products are formed. However, the actual area where transfection occurs is poorly defined, because cells may move in and out of the area imprinted with DNA during the time required until a phenotype can be observed. This obstacle may be overcome by combining reverse transfection with micro patterning.
Materials and methods
Preparation of surfaces
PVA was covalently grafted onto the surface of glutaraldehyde-activated glass cover slips, followed by heat annealing of the PVA film. Precleaned cover slips were immersed for 30 min in ethanol containing 2% (3-aminopropyl)trimethoxysilane (Sigma) and 1% acetic acid, washed with acetone and ethanol, and air-dried. The surfaces were activated by immersion in a solution of 2.5% glutaraldehyde in 0.015 M phosphate-buffered saline (PBS; pH 7.2) for 2 h, washed with water and air-dried. A 5% (w/v) solution of PVA (Mr 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–70000; Sigma, Vienna, Austria) in sterile water was evenly distributed over the cover slips (7.5 µL cm−2), incubated at 130 °C for 2 h, washed with ethanol and water, and air-dried. In some experiments, PVA was immobilized by heat annealing alone,15 omitting silanization and activation with glutaraldehyde. Corning® Ultra Low Attachment dishes (ULAs) were obtained from Corning Life Sciences (Schiphol-Rijk, The Netherlands).
000–70000; Sigma, Vienna, Austria) in sterile water was evenly distributed over the cover slips (7.5 µL cm−2), incubated at 130 °C for 2 h, washed with ethanol and water, and air-dried. In some experiments, PVA was immobilized by heat annealing alone,15 omitting silanization and activation with glutaraldehyde. Corning® Ultra Low Attachment dishes (ULAs) were obtained from Corning Life Sciences (Schiphol-Rijk, The Netherlands).
Surface patterning
For chemical surface patterning, dilute aqueous solutions of NaOCl (0.5% unless otherwise stated) were spotted onto the dry surfaces using robot-controlled stainless steel pins with a diameter of 0.32 or 0.64 mm, or borosilicate glass pipettes with a tip diameter of approximately 50 µm, which were prepared using a DMZ Universal Puller (Zeitz Instrumente, München, Germany). After drying in a tissue culture hood, the surfaces were throroughly washed with 1 M HCl, PBS and sterile water. The samples were used immediately or stored dry without special provisions.For modification by UV light, the surfaces were irradiated for 15 min in a reactive atmosphere through a contact mask using a Xe2*-excimer lamp (Heraeus-Noblelight, Hanau, Germany). The lamp emits light with a centre wavelength of 172 nm, a spectral bandwith of about 16 nm, and an intensity of approximately 20 mW cm−2. During irradiation, the surfaces were flushed with air at a pressure of 5 mbar.
Cell culture
Rat skeletal myoblasts16 were maintained at 37 °C in Dulbecco's Modified Eagle Medium (DMEM; EuroClone, Pero, Italy) supplemented with 20% fetal bovine serum (FBS; EuroClone), 10 µg mL−1 insulin (Sigma) and 5 µg mL−1 gentamycin (EuroClone) in a humidified atmosphere containing 5% CO2. Human embryonic kidney cells (HEK 293) were maintained in DMEM/F12 (EuroClone) supplemented with 10% FBS and 5 µg mL−1 gentamycin. Colon carcinoma cells (Caco-2) were maintained in Minimum Essential Medium (MEM; Sigma) supplemented with 10% FBS and 5 µg mL−1 gentamycin. Cells were dissociated with PBS containing 0.05% trypsin and 0.02% EDTA and counted with a hemacytometer. Population doubling times were calculated by the following equation:| tg = (log2 ×
t)/(logN
− logN0) |
Cells were seeded onto micropatterned surfaces (at a density of approximately 5000 cells cm−2, unless otherwise indicated) and allowed to attach. Floating cells were removed by washing the surfaces once or twice with PBS.
Reverse transfection
Plasmid DNA (pGFP37) encoding a modified green fluorescent protein17 (GFP) was purified from Escherichia coli using a Midi Prep Plasmid kit (Qiagen, Hilden, Germany). Transfection mixtures were prepared by mixing 0.5 µL of plasmid DNA solution (0.5 µg DNA) with 6.25 µL of EC buffer (Effectene Transfection kit, Qiagen), 2 µL Enhancer solution and 2.5 µL of the Effectene reagent. After a 15 min incubation at room temperature, 1.25 µL of 1 M sucrose and 35 µL of 0.1% gelatin solution (Sigma) were added.For fabrication of transfection arrays, 0.5% NaOCl was spotted on cover slips covalently coated with PVA. Residual NaOCl was removed by washing the cover slips as described above. The cover slips were briefly dried in a laminar flow hood and re-installed in the spotting instrument. Then, the freshly prepared DNA mixtures were spotted onto the areas previously treated with NaOCl. For this procedure, the cover slips were mounted in tightly fitting holders to permit spotting of the different solutions onto identical positions. As negative controls, transfection mixtures lacking DNA were spotted adjacent to spots with DNA. HEK 293 cells were suspended in the standard medium (containing 10% FBS) and were seeded on the array at a density of approximately 25000 cells cm−2. GFP expression was assayed after 2 days using fluorescence microscopy.
Microscopy and image analysis
Phase contrast microscopic images were captured using either an AxioCam HR (Zeiss, Oberkochen, Germany) or a CC-12 (Olympus, Hamburg, Germany) CCD camera and analysed using the image analysis software ImageJ v. 1.35 (National Institutes of Health; download available at http://rsb.info.nih.gov/ij/). Cell number and the area covered by cells was estimated after conversion into binary images, using procedures similar to those described by Endler et al.18 Fluorescence images were acquired with a Zeiss LSM 510 inverted laser scanning microscope equipped with a transmitted light detector for acquisition of corresponding phase contrast images.Harvesting of cell colonies
Single cell colonies from patterened areas were harvested under microscopic control utilising a robotic system for automated isolation of cell colonies (CellCelector, Aviso, Greiz-Gommla, Germany). The instrument consists of an inverted microscope equipped with a CCD camera and a motorised stage holding the cell culture dish, a high-precision robotic arm, several racks for consumables and a heated holder for a reagent vial. The robotic arm is equipped with a conical receiver for a tip of an air-displacement pipette, which is connected via a tube to a 50 µL syringe with a motorised plunger. The entire unit is housed in a laminar flow hood providing a sterile atmosphere and is controlled by a personal computer. For a harvesting cycle, a 20 µL pipette tip is taken up by the robotic arm and filled with 10 µL of pre-warmed trypsin solution. Then, the tip is inserted in a Clonetip® (a miniaturized, disposable cloning cylinder with an inner diameter of 1 or 2 mm; Brand, Wertheim, Germany). A schematic representation of the tip assembly is shown in Fig. 6. The assembly is moved over a cell colony and lowered onto the surface, thereby enclosing the cell colony. The chamber is sealed by a silicon ring attached to the bottom of the Clonetip. The trypsin solution is dispensed within the Clonetip by several plunger strokes. After detachment of the cells from the surface, the cells are dispensed by plunger strokes, aspirated into the pipette tip and delivered into a multiwell plate pre-filled with nutrient medium. Finally, the tip assembly is disposed into a waste station. The system can be used to scan an entire dish, to identify cell colonies by automated image analysis and to store the positions of cell colonies for subsequent harvests, or can be programmed to harvest colonies from pre-defined positions.Results and discussion
Preparation and properties of cell-repellent surfaces
PVA was covalently bonded on aminosilane-derived, glutaraldehyde-activated glass cover slips, followed by thermal immobilization of the PVA film.19 PVA can be immobilized on glass by heat-annealing alone.15 However, in our hands non-covalently bonded films were less durable and required careful handling, because small scratches occurring during routine manipulations frequently resulted in detachment of the entire coating. As an alternative to glass as the support material, we used polystyrene plates coated with a neutrally charged hydrogel (ULAs), which are commercially available in a range of formats (the chemical structure of the hydrogel is not disclosed by the manufacturer). When rat skeletal myoblasts, HEK 293 or Caco-2 cells were seeded onto the surfaces, virtually no attachment and spreading of cells was observed. The cells remained round, occasionally forming aggregates after a few hours of incubation. With the exception of Caco-2 cells, which tended to stick to the surface, the cells and aggregates could be removed by gentle washing with medium or PBS. Similar observations have been reported for NIH3T3 fibroblasts,20 embryonic rat cerebral cortical stem cells,21 HT29 human colon adenocarcinoma cells15 and ARPE-19 human retinal pigment epithelial cells22 seeded on PVA gels, and for HT29 cells,15 monocytes and macrophages23 seeded on ULAs. Taken together, these results indicate that both surfaces resist the attachment of a wide range of mammalian cells.Micro patterning
For chemical surface patterning, NaOCl solutions were spotted onto the surfaces and allowed to dry. Before seeding cells onto the surface, residual NaOCl and caustic soda were removed by washing the entire surface with acid and PBS. Rat skeletal myoblasts, HEK 293 and Caco-2 cells adhered and spread exclusively on treated areas (Fig. 1). The only exceptions were Caco-2 cells growing on modified PVA films (Fig. 1f). A high proportion of these cells remained roundish, indicating insufficient attachment. | ||
| Fig. 1 Phase contrast images of mammalian cells growing for 3 days on ULAs (a–c) and PVA films covalently bonded on glass cover slips (d–f) chemically modified with 0.5% NaOCl. (a, d) Rat skeletal myoblasts. (b, e) HEK 293 cells. (c, f) Caco-2 colon carcinoma cells. Scale bar: 200 µm. | ||
Solutions containing as little as 0.1% NaOCl were sufficient to generate adhesive islands (Fig. 2). We routinely use 0.5% NaOCl on PVA coatings as well as on ULAs. Since the creation of adhesive areas requires nothing more than spotting of a single solution, it should be possible to employ various technical options, including a standard microarray spotter, microcontact printing, ink jet printers24 or precision spraying.25 The effective NaOCl concentration and reaction time on a spot depends on the rate of evaporation of the solute during transfer onto the surface, spot size and relative humidity during the spotting process. Hence, other spotting techniques may require optimization of the NaOCl concentration. Applications are not restricted to circular spots; using borosilicate glass pipettes connected to a motorised micromanipulator, we created line patterns. When rat skeletal myoblasts were seeded on these patterns, the cells aligned (Fig. 3) and upon serum deprivation, they fused into multinucleate myotubes (data not shown). Time-lapse video microscopic analysis revealed that the myoblasts spread on the treated areas, but lamellipodia extended only up to the limiting boundary (see Supplemental movie M1†). Occasionally, cell processes extended beyond the adhesive area, but were quickly retracted. Time-lapse microscopy also revealed that dividing cells were only weakly attached, displaying a roundish appearance. However, these cells flattened and attached firmly immediately after cell division.
 | ||
| Fig. 2 Effect of the NaOCl concentration on attachment of rat skeletal myoblasts on chemically modified ULAs. NaOCl spots with a diameter of approximately 0.75 mm (0.44 mm2) were generated using a stainless steel pin. The area covered by cells was estimated after 3 days by digital image analysis of phase contrast microscopic images. Weakly attached and floating cells were removed by washing the plate before image acquisition. The data represent the mean ± SD (n = 5). | ||
 | ||
| Fig. 3 Growth of rat skeletal myoblasts on a line pattern. Cell-adhesive lines were generated on a ULA with 0.5% NaOCl. Phase contrast images were taken (a) 1 day and (b) 7 days after seeding the cells. Scale bar: 200 µm. | ||
Since fairly mild conditions were sufficient for chemical surface modification, we investigated whether the surfaces are amenable to photopatterning. We used vacuum UV light with a wavelength of 172 nm, which is able to induce bond-breakage and radical formation on polymer surfaces.12,26 Additionally, irradiation in air (under reduced pressure) results in effective production of ozone.27 The ozone reacts with excited groups in the polymer, leading to the formation of polar oxidic groups and etching. These effects may be similar to those caused by exposure of the polymer surfaces to the bleaching agent NaOCl. Localized modification was achieved through irradiation of the samples through a contact mask. Upon seeding onto ULA surfaces modified by irradiation, cells attached almost exclusively on the exposed areas, yielding islands of cells with a very similar microscopic appearance to those created by NaOCl treatment (Fig. 4). With our experimental setup, an irradiation time of 15 min was necessary to create cell-adhesive domains. When samples were irradiated for 5 min, only a few, roundish cells attached to the exposed areas (data not shown).
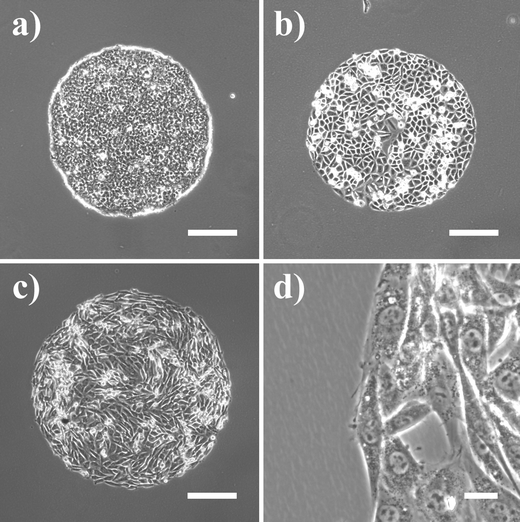 | ||
| Fig. 4 Photopatterning of a hydrogel. Circular adhesive areas were created by exposure of ULAs to UV light emitted by a Xe2*-excimer lamp through contact masks. (a) HEK 293 cells. (b) Caco-2 cells. (c) Rat skeletal myoblasts. (d) Higher magnification image of rat skeletal myoblasts growing at the border of an irradiated spot. Scale bar (a–c): 200 µm; (d): 20 µm. | ||
The patterned surfaces were remarkably stable. They could be re-used after the removal of cells by trypsin treatment and sterilization with 70% ethanol (20 min) without apparent loss in pattern fidelity. PVA-based surfaces could be maintained for at least 3 weeks under cell culture conditions, and ULAs for at least 9 weeks, exceeding the shelf life of many other micropatterned surfaces reported in the literature.28 Although UV light can be utilised for patterning, we found no indication that room light or illumination during routine fluorescence microscopy negatively affects pattern fidelity. Pure PVA shows no absorption of light with wavelengths above 250 nm.29 We verified this spectral characteristic for PVA immobilized on glass by spectroscopic ellipsometry (unpublished observation).
Since PVA coatings have been shown to exhibit excellent long-term performance in devices for capillary and chip electrophoresis,19,30 the techniques outlined here could be particularly useful for the creation of cell-adhesive areas in re-usable microfluidic cell culture devices. Apart from glass as the support material, PVA may be grafted onto a variety of polymers, provided that the material is able to withstand the heat annealing step. Surface patterning by chemical modification is exceptionally simple and rapid, but the spatial resolution achievable is limited by the technologies available to deposit a liquid. For the creation of complex patterns on the micrometre scale, treatment of the surfaces by UV projection patterning may be the method of choice.
Cell proliferation and harvesting of cell colonies
To compare the proliferation of rat skeletal myoblasts on chemically modified ULAs with that of myoblasts growing on tissue culture grade polystyrene (TCPS), the entire surface of ULAs was treated with 0.5% NaOCl. Cells were seeded onto the surfaces and population doubling times were estimated after 3 days by counting the cells harvested by trypsin treatment. Doubling times of cultures on modified ULAs (29.7 ± 4.7 h, n = 4) were not significantly different from those of cultures on TCPS (28.5 ± 2.2 h). To assess proliferation rates of cells growing on spots, cells were seeded at a very low density (approximately 50 cells cm−2) on a ULA containing an array of 100 NaOCl spots with a diameter of approximately 0.75 mm and a center-to-center distance of 1 mm. One day after seeding, a total of 90 cells were detected on 39 spots, each containing 1 (8 spots) to 9 (1 spot) cells. Their proliferation was monitored over a period of 4 days using a motorised, programmable microscopic stage in combination with automated image acquisition (Fig. 5). The entire population had a doubling time of 27.4 h. These results suggest that the growth characteristics of the cells on spots are not different from those cultured on TCPS. | ||
| Fig. 5 Dynamic monitoring of the proliferation of cells on a chemically modified ULA. Rat skeletal myoblasts were seeded at a low density on an array of 100 spots. Cell number per spot was monitored by automated image acquistion. The data from 25 spots with cells proliferating throughout the experiment are shown. | ||
We successfully harvested cell colonies from chemically patterned surfaces using an automated device (CellCelector, Aviso). By means of a robotic arm, a miniaturized cloning cylinder (Clonetip) fitted onto a pipette tip prefilled with trypsin solution was placed over a colony. The encircled area (with a diameter of 1 or 2 mm, depending on the type of Clonetip used) was perfused with the trypsin solution. Dissociated cells were aspirated and delivered into a multiwell plate (Fig. 6). Since the tip assembly was placed over cells submerged in medium, a more concentrated trypsin solution (0.1–0.2% trypsin and 0.04% EDTA, prewarmed to 39 °C) was used to compensate for the dilution of trypsin within the chamber. Before harvests, the nutrient medium in the culture dishes (containing serum, which would inactivate trypsin) was replaced by PBS. The time required to harvest a colony and harvest efficiencies varied, depending on cell type and density. In the case of rather weakly attaching cells such as HEK 293, 70–90% of the cells of a colony could be harvested within 2–3 min.
 | ||
| Fig. 6 Harvesting of a rat skeletal muscle cell colony growing on a chemically modified ULA. (a) Schematic representation of the harvesting process. Prior to a harvest, a robotic arm takes up a pipette tip. The pipette tip is filled with trypsin solution by means of the syringe attached to the robotic arm. The arm then inserts the pipette tip into a cylindrical Clonetip. The assembly is placed over a cell colony and trypsin is delivered into the perfusion chamber formed by the Clonetip. After detachment, the cells are aspirated into the pipette tip. Phase contrast images of the harvesting process (b–e). (b) Colony before harvesting. (c) During harvest, the Clonetip is in place over the colony; light entering the enclosed chamber through the pipette tip allows monitoring of the detachment of cells. (d) Area of the colony after harvest of the cells. (e) Harvested cells after delivery into a 12-well plate and attachment. Scale bar: 200 µm. | ||
The experimental design outlined above could potentially provide a new tool for cell cloning. Since migration of cells is restricted, seeding cells at a low density on a high-density array of spots increases the likelihood that a colony originates from a single cell. Combined with automated image acquisition, the platform allows the monitoring of cell number per spot, the proliferation rates of the cells and other properties (such as fluorescence). Based on these data, candidate colonies can be pre-selected for harvesting.
Patterned reverse transfection
Chemically patterned PVA films were evaluated for their ability to support substrate-mediated gene delivery. As a model system, we used a plasmid encoding GFP and HEK 293 cells, following the original protocol for reverse transfection14 as modified by Baghdoyan et al.31 Mixtures of DNA with transfection reagents were spotted onto areas which had previously been made cell-adhesive by treatment with NaOCl. After seeding the cells onto the array, expression of GFP was only detected over spots loaded with DNA, but not on adjacent control spots carrying transfection mixtures without DNA (Fig. 7). Transfection efficiencies for this system were comparable to those reported elsewhere.31 This and other studies32 demonstrate that it is possible to combine micro patterning of cells with local transfection. The combined system facilitates the use of light microscopy for spot identification and permits quantification of cell number, transfection efficiency and detection of cytotoxic effects of the expressed protein (or of any of the transfection reagents), undisturbed from cells invading the spotted area. | ||
| Fig. 7 Patterned reverse transfection of HEK 293 cells with a plasmid encoding GFP. DNA was printed onto NaOCl spots on a PVA film covalently bonded on to glass. (a) Fluorescence image of a field covering four spots. Transfection was confined to the spots imprinted with plasmid. Scale bar: 200 µm. (b) Merged fluorescence and phase contrast images. (c) Higher magnification image of a single spot loaded with DNA. Scale bar: 100 µm. | ||
Conclusions
This study demonstrates simple and inexpensive techniques for micro patterning of cell-repellent hydrogels. Patterns can be created by single-step procedures, either by spotting dilute solutions of NaOCl or by exposure to UV light. Selective cell adhesion and growth was demonstrated for three types of mammalian cells. The surfaces are exceptionally durable under cell culture conditions. Since no proteins are required to render domains cell-adhesive, the patterned surfaces can be re-used after the removal of cells by protease treatment. We show that micro patterning can be combined with reverse transfection, extending the capabilities of live cell microarrays. Furthermore, we envisage patterned surfaces as a new tool for the isolation of individual cell clones, combined with the possibility of tracking individual cell fates (or phenotypes) prior to harvesting.Acknowledgements
We thank Dr Annette Hohaus (University of Vienna, Department of Pharmacology and Toxicology) for providing plasmid DNA. This work was supported by grant N0102-NAN (NSI-NBPF), of the Austrian NANO Initiative.References
- K. K. Parker, A. L. Brock, C. Brangwynne, R. J. Mannix, N. Wang, E. Ostuni, N. A. Geisse, J. C. Adams, G. M. Whitesides and D. E. Ingber, FASEB J., 2002, 16, 1195–1204 CrossRef CAS.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS.
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425–1428 CrossRef CAS.
- C. J. Flaim, S. Chien and S. N. Bhatia, Nat. Methods, 2005, 2, 119–125 CrossRef CAS.
- A. Tourovskaia, X. Figueroa-Masot and A. Folch, Lab Chip, 2005, 5, 14–19 RSC.
- R. Singhvi, A. Kumar, G. P. Lopez, G. N. Stephanopoulos, D. I. Wang, G. M. Whitesides and D. E. Ingber, Science, 1994, 264, 696–698 CrossRef CAS.
- A. J. Engler, M. A. Griffin, S. Sen, C. G. Bonnemann, H. L. Sweeney and D. E. Discher, J. Cell Biol., 2004, 166, 877–887 CrossRef CAS.
- V. A. Liu, W. E. Jastromb and S. N. Bhatia, J. Biomed. Mater. Res., 2002, 60, 126–134 CrossRef CAS.
- Y. C. Wang and C.-C. Ho, FASEB J., 2004, 18, 525–527 CAS.
- S. Iwanaga, Y. Akiyama, A. Kikuchi, M. Yamato, K. Sakai and T. Okano, Biomaterials, 2005, 26, 5395–5404 CrossRef CAS.
- E. Detrait, J. B. Lhoest, B. Knoops, P. Bertrand and P. van den Bosch de Aguilar, J. Neurosci. Methods, 1998, 84, 193–204 CrossRef CAS.
- R. Mikulikova, S. Moritz, T. Gumpenberger, M. Olbrich, C. Romanin, L. Bacakova, V. Svorcik and J. Heitz, Biomaterials, 2005, 26, 5572–5580 CrossRef CAS.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- J. Ziauddin and D. M. Sabatini, Nature, 2001, 411, 107–110 CrossRef.
- S. N. Krylov and N. J. Dovichi, Electrophoresis, 2000, 21, 767–773 CrossRef CAS.
- H. C. Ott, S. Berjukow, R. Marksteiner, E. Margreiter, G. Bock, G. Laufer and S. Hering, J. Physiol., 2004, 558, 793–805 CAS.
- M. Grabner, R. T. Dirksen and K. G. Beam, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1903–1908 CrossRef CAS.
- E. E. Endler, P. F. Nealey and J. Yin, J. Biomed. Mater. Res., Part A, 2005, 74, 92–103 Search PubMed.
- D. Belder, A. Deege, F. Kohler and M. Ludwig, Electrophoresis, 2002, 23, 3567–3573 CrossRef CAS.
- C. R. Nuttelman, D. J. Mortisen, S. M. Henry and K. S. Anseth, J. Biomed. Mater. Res., 2001, 57, 217–223 CrossRef CAS.
- T.-H. Young and C.-H. Hung, Biomaterials, 2005, 26, 4291–4299 CrossRef CAS.
- C. J. Lee, M. S. Blumenkranz, H. A. Fishman and S. F. Bent, Langmuir, 2004, 20, 4155–4161 CrossRef CAS.
- M. Shen and T. A. Horbett, J. Biomed. Mater. Res., 2001, 57, 336–345 CrossRef CAS.
- E. A. Roth, T. Xu, M. Das, C. Gregory, J. J. Hickman and T. Boland, Biomaterials, 2004, 25, 3707–3715 CrossRef CAS.
- M. N. De Silva, J. Paulsen, M. J. Renn and D. J. Odde, Biotechnol. Bioeng., 2006, 93, 919–927 CrossRef.
- J. Heitz, T. Gumpenberger, H. Kahr and C. Romanin, Biotechnol. Appl. Biochem., 2004, 39, 59–69 CrossRef CAS.
- J. N. Pitts and J. G. Calvert, in Photochemistry, John Wiley & Sons, Hoboken, New Jersey, 1967 Search PubMed.
- C. M. Nelson, S. Raghavan, J. L. Tan and C. S. Chen, Langmuir, 2003, 19, 1493–1499 CrossRef CAS.
- M. Matsumoto, K. Imai and Y. Kazusa, J. Polym. Sci., 1958, 117, 426–428 CrossRef.
- D. Belder, A. Deege, H. Husmann, F. Kohler and M. Ludwig, Electrophoresis, 2001, 22, 3813–3818 CrossRef CAS.
- S. Baghdoyan, Y. Roupioz, A. Pitaval, D. Castel, E. Khomyakova, A. Papine, F. Soussaline and X. Gidrol, Nucleic Acids Res., 2004, 32, e77 CrossRef.
- A. K. Pannier, B. C. Anderson and L. D. Shea, Acta Biomater., 2005, 1, 511–522 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental movie M1 with description. See DOI: 10.1039/b601803c |
| This journal is © The Royal Society of Chemistry 2006 |