High performance computing in materials chemistry
Abstract
This issue of Journal of Materials Chemistry focuses on recent developments in the use of high performance computing for contemporary modelling studies in materials chemistry. Richard Catlow and Scott M. Woodley, Guest Editors, introduce the issue and highlight the key aspects of the field.
Computer modelling techniques are now influencing almost all areas of chemistry, but there is no sub-discipline in which they have had a greater impact than materials chemistry. Simulation techniques are now used routinely to generate accurate models of the structures of crystalline and amorphous solids, and to study surfaces and defects of complex materials and their properties;1 while the challenge of modelling properties and problems related to synthesis and reactivity is being met by increasingly detailed and realistic simulations.
The rapid growth of the field has been enabled not only by developments in theory, algorithms and software, but also by the continuing exponential growth in the power of computer hardware. Two factors contribute to the latter: first, the improvement in the processing power from the increase in processing speed and the amount and speed of the available memory of the individual processors; secondly, the growing ability to exploit massive parallelism, in which computational tasks are distributed over large numbers (often >1000) of processors. Scientific computing is indeed now a pyramid, where the base represents the many desktop computers (which now have considerably greater power than the “supercomputers” of the 1980s) and at the apex are “high performance” computers (HPC), i.e. the systems with the greatest processing power and available memory; although today's HPC very rapidly become tomorrow's mid range facility. For example, important components, like speed and number of processors, memory and electrical switches, of the UK's national high-performance computing service, namely HPCx (currently 1536 processors, delivering 9.2 TeraFlop s−1 peak, equipped with 3.2 TeraByte of memory and 36 TeraByte of disk), is continually upgraded over its planned lifetime of six years.
Computational materials chemistry exploits the full range of current hardware, and indeed desktop resources are now entirely adequate for a large number of studies in, for example, structure and defect modelling. The articles in this theme issue concentrate, however, on the opportunities in high performance computing, which is leading to unprecedented realism and accuracy in modelling complex systems.
The papers in this issue illustrate many of the major themes in contemporary modelling studies in materials chemistry. Modelling of complex structures is illustrated by the work of Tilocca and de Leeuw, where Carr–Parrinello techniques are used in accurate prediction of the structure and electronic properties of silicate glasses (Fig. 1). The topical theme of nanochemistry is explored in the paper of Woodley et al., in which the structure, energies and electronic properties of transition metal nanoparticles are modelled using Density Functional Theory (DFT) techniques; while the paper of Tomić et al. examines the electronic structures of zinc blend semiconductor quantum dots. Different aspects of nanochemistry are examined in the work of Tay and Bresme, that models the property of water about passivated gold nanoparticles, which are approximately 3 nm in diameter (see Fig. 2).
 | ||
| Fig. 1 From Tilocca and de Leeuw.2 | ||
 | ||
| Fig. 2 From Tay and Bresme.3 | ||
The interrelated themes of surface chemistry and reactivity are the subject of several contributions. Coquet et al. uses the recently developed O(N) DFT methods to model the adsorption of gold on MgO (see Fig. 3). The problem of diffusion and adsorption of H2 on Pt and PtSn—key systems in catalytic science—is investigated by Fearon and Watson. In the contribution of To et al., embedded cluster (QM/MM) models are used to refine models of the active sites and mechanisms of their formations in the TS-1 oxidation catalyst, while Wander et al. again exploit DFT methods for the study of the reactivity of halide surfaces.
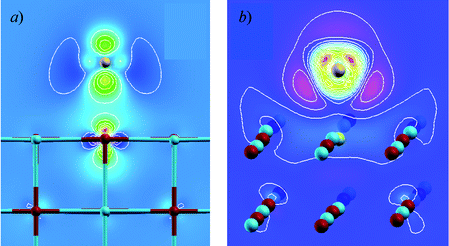 | ||
| Fig. 3 From Coquet et al. (Fig. 3).4 | ||
In contrast, atomistic simulations were deployed by Spagnoli et al. to provide new insight into the structures of aqueous solutions close to polar surfaces. The authors tackled the challenging problem of surface chemistry, which required large-scale molecular dynamics simulations.
The properties of defective and doped solids is an enduring theme in materials chemistry, and one which is illustrated in two articles: first, the work of Koudriachova and Harrison on doped TiO2 (Fig. 4), and second, the detailed survey of the defect chemistry of the mixed oxide FeSbO4 catalyst by Grau-Crespo et al.
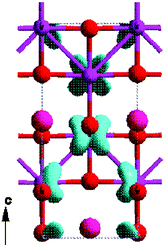 | ||
| Fig. 4 From Koudriachova and Harrison.5 | ||
The complexity of systems accessible to contemporary modelling studies is illustrated nicely by two articles: the work of Hawtin and Rodger on gas hydrate inhibitors (see Fig. 5), and the study of Kelly and Kantorovich on gas phase, nucleic acid-base superstructure (see Fig. 6).
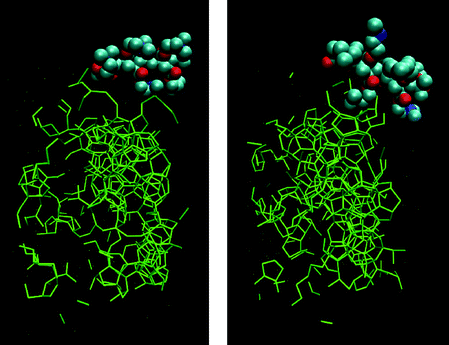 | ||
| Fig. 5 From Hawtin and Rodger (Fig. 10).6 | ||
 | ||
| Fig. 6 From Kelly and Kantorovich.7 | ||
Finally, the articles of Todorov et al. and of Plummer et al. demonstrate the key importance of code development and optimisation if the scientific potential of HPC is to be realised.
The articles in the issue all stem from the work of the UK's HPC Materials Chemistry Consortium (funded by EPSRC grants GR/S 13422 and EP/D504872), although they by no means represent an exhaustive list of research areas within their portfolio. This work made use of the facilities of HPCx, the UK's national high-performance computing service, which is provided by EPCC at the University of Edinburgh and by CCLRC Daresbury Laboratory, and funded by the Office of Science and Technology through EPSRC's High End Computing Programme. We would like to thank the staff within the HPCx User Administration and Helpdesk for their efficient and friendly help and advice. We are grateful to EPSRC for their support for this work, and we hope that the issue illustrates the range and impact of the materials chemistry which is enabled by HPC.
References
- R. Catlow, R. Bell, F. Cora, S. A. French, B. Slater and A. Sokol, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2005, 101, 513–547 RSC.
- A. Tilocca and N. H. de Leeuw, J. Mater. Chem., 2006 10.1039/b517362k.
- K. Tay and F. Bresme, J. Mater. Chem., 2006 10.1039/b600252h.
- R. Coquet, G. J. Hitchings, S. H. Taylor and D. J. Willock, J. Mater. Chem., 2006 10.1039/b601213b.
- M. V. Koudriachova and N. M. Harrison, J. Mater. Chem., 2006 10.1039/b600794p.
- R. W. Hawtin and P. M. Rodger, J. Mater. Chem., 2006 10.1039/b600285b.
- R. E. A. Kelly and L. N. Kantorovich, J. Mater. Chem., 2006 10.1039/b601364c.
| This journal is © The Royal Society of Chemistry 2006 |