Surface characterization and stability phenomena in Li2FeSiO4 studied by PES/XPS
Anton
Nytén
a,
Mårten
Stjerndahl
a,
Håkan
Rensmo
b,
Hans
Siegbahn
b,
Michel
Armand
c,
Torbjörn
Gustafsson
a,
Kristina
Edström
a and
John O.
Thomas
*a
aÅngström Advanced Battery Centre, Department of Materials Chemistry, Uppsala University, Box 538, SE-751 21 Uppsala, Sweden. E-mail: josh.thomas@mkem.uu.se
bDepartment of Physics, Ångström Laboratory, Uppsala University, Box 530, SE-751 21 Uppsala, Sweden
cLaboratoire de Réactivité et de Chimie des Solides, CNRS UMR 6007, Université de Picardie Jules Verne, 80039 Amiens Cedex 9, France
First published on 20th July 2006
Abstract
Photoelectron spectroscopy (PES) has been used to characterise the surface of Li2FeSiO4 cathodes extracted from lithium-ion batteries. Pristine, uncycled, air-exposed electrodes were first analysed and found to carry significantly greater amounts of Li2CO3 on their surfaces than electrodes stored under inert atmosphere. The surface film formed on electrochemical cycling of Li2FeSiO4 electrodes at 60 °C using a LiN(SO2CF3)2 salt based electrolyte revealed high salt stability and only small amounts of solvent reaction products. These were mainly of Li-carboxylate type; neither carbonates nor LiF were found. The excellent capacity retention (<3% over 120 cycles) and minimal irreversible capacity during the first cycle are probably a direct result of this very thin surface film. Li2FeSiO4 must therefore be seen as a most promising (and potentially cheap) positive electrode material for future large-scale Li-ion battery applications.
Introduction
A new group of electrochemically active silicate-based materials, Li2MSiO4 (M = Fe, Mn), has recently emerged as promising and potentially cheap cathode materials for large-scale Li-ion battery applications.1–3 The presence of strong Si–O bonds should promote the same lattice stabilization effect as in LiFePO4.4 Particularly Li2FeSiO4 has shown good reversibility on electrochemical cycling, with a loss of only ∼3% in capacity over more than 120 cycles. The theoretical capacity for Li2FeSiO4 is 166 mAh g−1 based on the reaction: Li2FeSiO4 → LiFeSiO4 + Li+ + e−. The shift observed in the potential plateau from 3.10 V to 2.80 V between the first and second cycle5 is suggested to be related to a structural rearrangement to a more stable structure. Although the details of this process are far from totally understood, it has been suggested that some of the Li ions (in the 4b site) and Fe ions (in the 2a site) interchange between the first and subsequent cycles.Surface-film formation at both the anode and cathode electrode/electrolyte interfaces is known to underlie irreversible capacity loss. Extensive studies of graphite anodes in a Li-ion battery have shown that a film is formed, the so-called Solid Electrolyte Interphase (SEI), during the first cycle, which totally covers the particle surface. This film contains different amounts of polymeric species, LiF, Li2CO3etc., depending on the lithium salt and organic solvent used in the electrolyte.6–9
Systematic studies of the surface film formed on transition-metal oxide cathodes and its influence on battery performance have also been made.10–14 These oxides have been shown to participate actively in the film formation.10,14 The resulting films comprise a mixture of mainly polycarbonates, LiF, LixPFy- and LixPOyFz-type compounds. The P-containing compounds result from the use of a LiPF6-based electrolyte. It has been shown that surface film formation is not only electrochemically driven; it can also have a chemical origin as a result of storage in the electrolyte.11–13 Cycling and storage at elevated temperature compared to room temperature have not resulted in any significant differences of the elemental composition of the surface film, but it is somewhat thicker and has a greater coverage.12 The increase observed in ac impedance measurements for LiNi0.8Co0.2O2 electrodes has been suggested to originate in the thicker surface layer formed at higher temperatures.15,16
However, from a study of electrochemically cycled LiFePO4 electrodes,17 it was concluded that no solvent-based reaction products were formed on the surface, indicating that the oxygen in the phosphate group does not take part in any such reactions, unlike the oxygen in the Co and Mn oxides. In LiFePO4, the surface film consisted only of different salt-based products. Depth profiling could show that this film did not completely cover the electrodes or, at least, was not thicker then the detection limit of a few nanometers.
The stability of pristine Li2FeSiO4 electrodes is here studied at ambient temperature by photoelectron spectroscopy (PES) using both synchrotron radiation (SR) and monochromatized AlKα radiation, and compared with that of electrodes cycled at 60 °C. The influence of cycling on surface-layer composition is discussed. To our knowledge, no previous PES study of this type has been made of a Li-ion battery cathode in contact with a LiTFSI-based electrolyte.
Experimental
Li2FeSiO4 was prepared by the solid-state reaction of Li2SiO3 with FeC2O4·2H2O. These starting materials were dispersed in acetone, mixed thoroughly and then ground together with 10 wt% polyethylene–poly(ethylene glycol). After evaporating the acetone, the mixture was heated to 700 °C for 20 h in a flow of CO–CO2 gas (50 : 50) to suppress the oxidation of Fe2+. The crystallographic structure was confirmed by X-ray powder diffraction (not shown here). The carbon content of the synthesised material was determined to be 4.68(5) wt% using carbon elemental analysis by the combustion method.5The material was cycled electrochemically using “coffee-bag” type cells. Electrodes were prepared by thinly spreading a mixture of 90% active material, 5% carbon black and 5% EPDM binder onto an aluminium foil; the thickness of the active layer is ca. 25 µm. Circular electrodes (3.14 cm2) with a typical loading of 3–5 mg of active material were dried under vacuum at 120 °C prior to assembly of the electrochemical cell. Half-cells of configuration <Li2FeSiO4 slurry | glass-wool separator soaked in electrolyte | Li foil> were assembled in an Ar-filled glove-box (<5 ppm H2O and O2). The electrolyte used was 1 M LiTFSI dissolved in ethylene carbonate (EC) and propylene carbonate (PC) (both Merck; battery grade) in a 1 : 1 ratio. The LiTFSI salt was also dried under vacuum at 120 °C prior to electrolyte mixing; the solvents were used as received. For the investigation of the stability phenomena, two electrodes were used (one air-exposed and one stored under an Ar atmosphere); an electrode stored in electrolyte for the same duration as the electrochemical cycling (3 days) was used as reference in the surface characterization.
Galvanostatic cycling was performed at 60 °C using a Digatron BTS 600 testing system. Cells were precycled three times at a C/20 rate (current density: 4.7 mA g−1) between 2.0 and 3.7 V, and then stored for one week at ambient temperature (under transport to the synchrotron light source). The cells were then dismantled in the glove-box and small pieces of the electrodes cut out, mounted on a sample holder, and transported to the PES equipment using a specially designed transport chamber to avoid contamination from air or moisture. All measurements were performed on unwashed electrodes to preserve any surface species formed during the cycling.
PES measurements using monochromatized AlKα at 1486.6 eV were performed on a PHI 5500 system. Two fresh electrodes, not exposed to electrolyte, were studied; one was exposed to air for a month, the other stored in an Ar-filled glove-box. Air exposure of maximum 2 h was unavoidable during the preparation of the electrodes. Absolute intensities for the different elements were used in the AlKα-excited spectra. The reported spectra for each element are scaled internally with respect to acquisition time, thus facilitating direct comparison within each element between the two samples. No charging effects (resulting from non-conducting surface species) were observed in the measurements. Peak assignments were made on the basis of measurements of the Li2FeSiO4 electrodes compared to reference compounds (Table 1),11,18–21 rather than using absolute binding energies.
| Assignments | Measured binding energy/eV | |||||||
|---|---|---|---|---|---|---|---|---|
| C1s | O1s | F1s | Si2p | S2p | N1s | Li1s | Fe2p | |
| a A carbon-treated Li2FeSiO4 electrode. b A carbon-black and EPDM binder electrode. | ||||||||
| Li2FeSiO4a | 284.4 | 530.5 | 101.4 | 709.5 | ||||
| Carbon blackb | 284.4 | |||||||
| Hydrocarbon | 285 | |||||||
| –COOR | ∼289 | 532.6 | ||||||
LiOC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)R O)R |
288 | ∼532.5 | ||||||
| LiOCH3 | 287.5 | 532.3 | ||||||
| Li2CO3 | 290 | 531.5 | 55 | |||||
| Li2O | 529 | 55 | ||||||
| LiF | 686 | ∼56.5 | ||||||
| LiN(SO2CF3)2 | 292.5 | 532.5 | 689.6 | 168.9 | 399.3 | |||
The SR-based PES measurements were performed on beam line I411 of the Swedish National Synchrotron Radiation Laboratory MAX.22 Measurements were made on a reference electrode (exposed to electrolyte) and on an electrochemically cycled electrode at excitation energies of about 780 eV. A background correction was applied to all spectra, and the reported binding energies were determined using the C1s carbon black and EPDM binder peak (284.4 eV) as reference. The high intensity of the synchrotron radiation means that it is important to verify that no radiation damage or charging effects influence the final result. Checks were made which confirmed that such effects can be neglected in the spectra shown here. Neither radiation damage nor charging effects are therefore a source of peak broadening.
It can be noted that no comparisons are made between elements; we simply comment qualitatively on what has not been formed during cycling, and state that the salt used appears to be stable. We do not wish to stretch our interpretation further at this stage, both due to the small quantities of material involved and the consequent relative uncertainty as to precisely what compounds are formed (see later).
Results and discussion
Electrochemical characterisation
Voltammograms for the first three cycles at 60 °C for carbon-coated Li2FeSiO4 are shown in Fig. 1. The reason for the observed shift in the potential plateau (from 3.10 V on the first charge cycle to 2.80 V on subsequent charge cycles) has been discussed in a recent paper, and will therefore not be addressed further here.5 The initial capacity of 120 mAh g−1 is constant over the first three cycles presented here. Indeed, the total capacity loss over 120 cycles is only ∼3%. The electrochemical cycling behaviour for an unexposed and an air-exposed sample is shown in Fig. 2. The low specific capacity for the air-exposed (13 mAh g−1) compared to the unexposed sample (113 mAh g−1) on the first charge cycle suggests that lithium is withdrawn from the structure on exposure to air. However, the capacity on the first discharge cycle is the same for the two samples. From complementary electrochemical measurements (not shown here) on cells with a graphite anode (instead of lithium), it was confirmed that the lithium withdrawn on exposure to air does not participate in the subsequent intercalation/deintercalation process. | ||
| Fig. 1 Voltammograms for the first three cycles of carbon-coated Li2FeSiO4 at 60 °C; cycle rate C/20 in 1 M LiTFSI EC : PC (1 : 1). Cycles 1, 2 and 3 are represented by solid, dashed and dotted curves, respectively. | ||
 | ||
| Fig. 2 First-cycle performance of (a) an unexposed and (b) an air-exposed sample of carbon-coated Li2FeSiO4 at 60 °C; cycle rate C/25 in 1 M LiTFSI EC : PC (1 : 1). | ||
Characterization of pristine unexposed and air-exposed Li2FeSiO4 electrodes
The PES spectra for the two electrodes are shown in Fig. 3. The C1s spectra contain two peaks: a major peak at 284.4 eV and a minor peak at 290.0 eV. The peak at 284.4 eV originates from the carbon black, EPDM binder and the carbon on the Li2FeSiO4 particles. The smaller peak is at the energy expected for carbonates, most likely in the form of Li2CO3 or possibly LiHCO3. Larger quantities of Li2CO3 were observed on the air-exposed compared to the unexposed electrode. Li2CO3 has been observed previously on the surface of pristine electrodes of transition-metal oxides like LiMn2O4, LiNiO2 and LiNi1−x−yCoxAlyO2,10,14,23 but was not observed on the surface of LiFePO4.17 The shoulder on the low-energy side of the carbonate peak in both spectra indicates the presence of less strongly bound carbon. A possible source could be some carboxyl compounds. | ||
| Fig. 3 PES spectra recorded using Al Kα radiation for pristine unexposed (left column) and air-exposed (right column) carbon-coated Li2FeSiO4 electrodes. Note: the intensities for each individual element are absolute, and have been normalised against measuring time to facilitate internal intensity comparison for each element. | ||
In the O1s spectra, a stronger signal at 531.5 eV for the air-exposed electrode, compared to the unexposed electrode, can be assigned to an increase of the amount of carbonate on the surface. A contribution from the oxygen in Li2FeSiO4 is clearly observed (at 530.5 eV) in the O1s spectrum for the unexposed electrode, whereas this contribution is smaller for the air-exposed electrode. This may be a result of the formation of carbonates on the Li2FeSiO4 surface on exposure to air, probably through the reaction of atmospheric oxygen and carbon dioxide with the lithium in Li2FeSiO4. Since the carbon black peak at 284.4 eV is not significantly weaker in the C1s spectrum for the air-exposed electrode, this would suggest that the coverage of the carbonate-based film is only partial. The peak in the Fe2p3/2 spectrum is composed of an Fe2+ component on the low binding-energy side and an Fe3+ component on the high binding-energy side. The shoulder on the high-energy side is clearly larger for the air-exposed electrode, indicating a larger amount of Fe3+ on the surface, probably from LiFe3+SiO4 as lithium is extracted. No major differences could be observed between the Si2p spectra for the two electrodes; the contribution is solely from the Si in the silicate groups. The Li1s peaks are asymmetric, indicating that the lithium near the surface exists in several states. It is suggested in reference 11 that the Li2CO3 peak should appear at 55 eV, implying more Li2CO3 at the surface of the air-exposed electrode, since the peak is narrower and more centred at 55 eV for the air-exposed electrode. This supports the conclusions drawn from the C1s and O1s spectra. No contribution from Li2CO3 could be detected in the XRD measurements, suggesting very small particle sizes (in the nm range). Indeed, the air-exposed sample showed no diffraction peaks whatsoever—not even from Li2FeSiO4. The major lesson here is that special care must be taken to minimize contact with air to prevent oxidation of Li2FeSiO4.
Characterization of uncycled, electrolyte-exposed and electrochemically cycled electrodes
PES spectra for electrochemically cycled Li2FeSiO4 electrodes are shown in Fig. 4. Only small differences can be seen between an electrode stored in electrolyte and an cycled electrode. For both electrodes, the main C1s peak (284.4 eV) derives from the carbon black, EPDM binder and the carbon coating on the Li2FeSiO4 particles. The peak at the highest binding energy (292.5 eV) originates from the LiTFSI salt. The absence of broadening or shift in this peak indicates that the salt is also present on the surface in its original form even after cycling. This is supported by the F1s spectrum, where the only peak present can be assigned to –CF3 (689.6 eV) in LiTFSI. Notably, a minimal amount of LiF (686 eV) is present on the surface of the two electrodes. The S2p and N1s spectra show only one major contribution—that from LiTFSI. Since no LiTFSI degradation products were detected on the surface, it can be concluded that the salt is stable under cycling.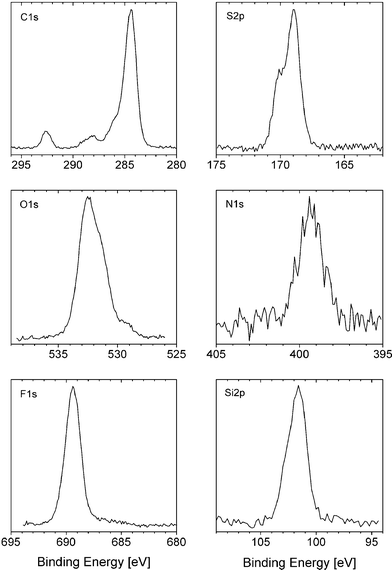 | ||
| Fig. 4 PES spectra recorded using synchrotron radiation (photon energy: 790 eV) for a carbon-coated Li2FeSiO4 electrode cycled at C/20 in 1 M LiTFSI EC : PC (1 : 1) at 60 °C. | ||
A small, rather broad peak in the C1s spectrum occurs around 288 eV (289 eV in the uncycled electrode) and a shoulder appears on the high binding-energy side of the main peak at 284.4 eV. Since no salt-based products are formed, these peaks must originate from solvent reaction products. Surface studies of LiMn2O4, LiNiO2 and LiNi0.8Co0.2O2 showed previously that the solvent reaction products were mainly hydrocarbons, polycarbonates and lithium alkyl carbonates.10,14 However, no peak is detected at 290–291 eV here, which implies the total absence of carbonate-based compounds. This also leads to the conclusion that the Li2CO3 initially present had already disappeared from the surface in the electrolyte-exposed electrode before electrochemical cycling.
The Li2CO3, which forms on pristine cathode materials such as LiNi0.8Co0.2O2 and LiNi0.8Co0.15Al0.05O2, is found to disappear on electrochemical cycling through reaction with the PF5 or HF from the LiPF6 salt used.14,24 That Li2CO3 could also be present here but completely covered by solvent reaction products, and therefore not detectable, is not realistic since Si2p peaks from Li2FeSiO4 are also detected after cycling.
In a surface study by Zhuang and Ross25 of the SEI layer formed on aged graphite electrodes, the dominant species were characterized to be lithium succinate (LiO2CCH2CH2CO2Li), lithium oxalate (Li2C2O4) and lithium methoxide (LiOCH3). They suggest lithium succinate as a model compound for lithium carboxylates such as Li formate, acetate and propionate. Such compounds could explain the presence of peaks at 286 and 288–289 eV in our C1s spectra. The carbon in LiOC(O)R is expected to have a peak at 288 eV, and the two carbon atoms adjacent to the two ester carbons in (−OC(O)CH2CH2(O)CO−) should give a peak at around 285.5 eV. The presence of some type of Li alkoxide (e.g., LiOCH3) is also possible. The peak in the C1s spectrum is here expected at ∼287.5 eV. These compounds would give rise to peaks in the range 531.5–532.5 eV in the O1s spectrum, which is at the boundary of our spectra. These types of surface species should accumulate on cycling at 60 °C.
The peak in the O1s spectrum is centred at 532.5 eV, with a shoulder on the low binding-energy side. A smaller contribution at 529 eV is also observed. Oxygen in LiTFSI is expected to show at 532.5 eV, and is most certainly the main contribution to this peak. The observed shoulder corresponds to a contribution from the silicate group (530.5 eV) and possibly also from some carbonyl or carboxylate oxygen in solvent reaction products. The peak at 529 eV can be assigned to Li2O.
The major part of the surface film would seem to originate from the salt in its original form, although more solvent reaction products are present after cycling compared to the reference electrode. However, the C1s peak from the carbon black, EPDM and the carbon coating is still the strongest for both electrodes, thus supporting the conclusion (also drawn for LiFePO4) that the surface film formed does not completely cover the electrode surface. The general character and composition of this surface layer on a cycled Li2FeSiO4 electrode is illustrated schematically in Fig. 5. The occurrence of solvent reaction products at the surface is in direct contrast with the situation found in LiFePO4.
 | ||
| Fig. 5 A schematic model of the surface layer components on a Li2FeSiO4 electrode cycled at C/20 in 1 M LiTFSI EC : PC (1 : 1) at 60 °C. | ||
Conclusions
The surface layer formed on Li2FeSiO4 has been investigated for the first time. Pristine electrodes exposed to air show larger amounts of carbonate-based compounds (Li2CO3 or LiHCO3) on their surfaces than electrodes stored under inert atmosphere. This indicates that lithium is withdrawn from the original structure on exposure to air. The LiTFSI salt used here is stable during electrochemical cycling, at least up to 3.7 V. Small amounts of solvent reaction products, possibly Li carboxylates, were found on the surface, but the main component of the surface film was LiTFSI salt in its original form. No LiF or carbonate-based compounds were found on the surface after electrochemical cycling. This suggests Li2FeSiO4 to be a highly promising and potentially cheap cathode material for use in large-scale Li-ion batteries.Acknowledgements
This work has been supported in Sweden by The Swedish Energy Agency (STEM), The Swedish Science Council (VR), The Swedish Foundation for Strategic Research (SSF) and The Knut and Alice Wallenberg Foundation (KAW); and, in part, by the EU-FP6 Network of Excellence (NoE) ALISTORE.References
- A. Abouimrane, N. Ravet, M. Armand, A. Nytén and J. O. Thomas, Abstract #350, presented at IMLB-12, Nara, Japan, 27 June–2 July 2004.
- A. Nytén, A. Abouimrane, M. Armand, T. Gustafsson and J. O. Thomas, Electrochem. Commun., 2005, 7, 156 CrossRef CAS.
- R. Dominko, M. Bele, M. Gaberšček, A. Meden, M. Remškar and J. Jamnik, Electrochem. Commun., 2006, 8, 217 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CAS.
- A. Nytén, S. Kamali, L. Häggström, T. Gustafsson and J. O. Thomas, J. Mater. Chem., 2006, 16, 2266 RSC.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CAS.
- A. M. Anderson, A. Henningsson, H. Siegbahn, U. Jansson and K. Edström, J. Power Sources, 2003, 119–121, 522 CrossRef CAS.
- A. M. Anderson and K. Edström, J. Electrochem. Soc., 2001, 148, A1100 CrossRef CAS.
- A. M. Anderson, M. Herstedt, A. Bishop and K. Edström, Electrochim. Acta, 2002, 47, 1885 CrossRef CAS.
- D. Aurbach, K. Gamolsky, B. Markovsky, G. Salitra, Y. Gofer, U. Heider, R. Oesten and M. Schmidt, J. Electrochem. Soc., 2000, 147, 1322 CrossRef CAS.
- T. Eriksson, A. M. Andersson, A. G. Bishop, C. Gejke, T. Gustafsson and J. O. Thomas, J. Electrochem. Soc., 2002, 149, A69 CrossRef CAS.
- T. Eriksson, A. M. Andersson, C. Gejke, T. Gustafsson and J. O. Thomas, Langmuir, 2002, 18, 3609 CrossRef CAS.
- G. Pistoia, A. Antonini, R. Rosati and D. Zane, Electrochim. Acta, 1996, 41, 2683 CrossRef CAS.
- A. M. Andersson, D. P. Abraham, R. Haasch, S. MacLaren, J. Liu and K. Amine, J. Electrochem. Soc., 2002, 149, A1358 CrossRef CAS.
- C. H. Chen, J. Liu and K. Amine, J. Power Sources, 2001, 96, 321 CrossRef CAS.
- D. P. Abraham, J. Liu, C. H. Chen, Y. E. Hyung, M. Stoll, N. Elsen, S. MacLaren, R. Twesten, R. Haasch, E. Sammann, I. Petrov, K. Amine and G. Henriksen, J. Power Sources, 2003, 119–121, 511 CrossRef CAS.
- M. Herstedt, M. Stjerndahl, A. Nytén, T. Gustafsson, H. Rensmo, H. Siegbahn, N. Ravet, M. Armand, J. O. Thomas and K. Edström, Electrochem. Solid-State Lett., 2003, 6, A202 CrossRef CAS.
- M. Herstedt, D. P. Abraham, J. B. Kerr and K. Edström, Electrochim. Acta, 2004, 49, 5097 CrossRef CAS.
- H. Ota, Y. Sakata, X. Wang, J. Sasahara and E. Yasukawa, J. Electrochem. Soc., 2004, 151, A437 CrossRef CAS.
- N. M. D. Brown, J. A. Hewitt and B. J. Meenan, Surf. Interface Anal., 1992, 18, 187 CAS.
- L. J. Rendek, G. S. Chottiner and D. A. Scherson, J. Electrochem. Soc., 2002, 149, E408 CrossRef CAS.
- M. Bässler, J. O. Forsell, O. Björneholm, R. Feifel, M. Jurvansuu, S. Aksela, S. Sundin, S. L. Sorensen, R. Nyholm, A. Ausmees and S. Svensson, J. Electron Spectrosc. Relat. Phenom., 1999, 101–103, 953 CrossRef CAS.
- K. Matsumoto, R. Kuzuo, K. Takeya and A. Yamanaka, J. Power Sources, 1999, 81–82, 558 CrossRef CAS.
- S.-W. Song, G. V. Zhuang and P. N. Ross, Jr., J. Electrochem. Soc., 2004, 151, A1162 CrossRef CAS.
- G. V. Zhuang and P. N. Ross, Jr., Electrochem. Solid-State Lett., 2003, 6, A136 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |