Blood substitutes: from chemistry to clinic
Kenneth C.
Lowe
School of Biology, Faculty of Medicine and Health Sciences, University of Nottingham, University Park, Nottingham, UK NG7 2RD
First published on 21st August 2006
Abstract
Blood substitutes are oxygen-carrying fluids that aim to provide an alternative to the transfusion of blood. Strategies for developing such substitutes have involved the production of materials based on the naturally occurring respiratory pigment, haemoglobin (Hb), or synthetic, chemically inert, fluorinated liquids called perfluorochemicals (PFCs). Commercial products in both categories have been developed and some approved for clinical use, primarily to facilitate oxygen supply to tissues during surgery or therapy. The latest research is focused on using microbial and plant ‘cell factories’ to express recombinant Hb, understanding the properties of polymeric Hbs from invertebrate animals, and the use of feedback from stakeholders to inform the development of new educational materials to assist patients to make informed choices on future transfusion options.
 Kenneth Lowe | Kenneth Lowe received his B.Sc. (Zoology) from the University of Leeds in 1976 and Ph.D. (Physiology) from the University of Cambridge in 1980. He is currently Associate Professor and Reader in Biotechnology at the University of Nottingham. His principal research interests are in developing ‘blood substitutes’ and he is currently Scientific Co-ordinator of a multi-national, EU-funded research programme, The ‘EuroBloodSubstitutes’ Project (www.eurobloodsubstitutes.com). |
Introduction
Blood transfusion has its origins in the 1660s when the English physician, Richard Lower, and later the Frenchman, Jean-Baptiste Denis, began the first transfusions of blood, initially from animal to animal, and subsequently from animals to humans.1 In December 1818, James Blundell performed the first transfusion from one human patient to another, thereby laying the foundations for the dramatic refinements in transfusion practice and the establishment of blood banks which occurred in the 20th century.Modern blood transfusion in industrialised countries is a universally practised and remarkably safe clinical procedure. Blood or concentrated (‘packed’) red cells are routinely transfused following extensive blood loss or as a treatment for chronic anaemia. In both situations, the aim of the transfusion is to not only increase blood volume but also increase the arterial oxygen content thereby enhancing the delivery of oxygen to the tissues. Blood transfusion is not, however, a zero-risk intervention and there are several physiological and practical problems associated with this procedure. Allogeneic (donor) blood transfusion may cause fatal haemolytic reactions and compromise immune system function in the recipient. Such effects, albeit rare, together with the need for sophisticated and expensive blood processing, storage and cross matching, coupled with increasing public concerns about the safety of blood products,2–5 have driven the on-going search for safe and effective substitutes. The useful shelf life of donated blood when stored at 4 °C in a refrigerator is only ∼42 days (35 days in the UK), even with the use of modern nutrient additives, such as mannitol, glucose and adenine. Inevitably, some blood collected from donors is wasted.
Concerns about blood safety are illustrated by the UK Blood Services' decision in 1998 to suspend the fractionation of UK plasma and to introduce routine filtration of blood to remove white blood cells (leucodepletion). These precautions were introduced to minimise the risk of transmitting the prions causing human variant Creutzfeldt–Jakob Disease (vCJD) through transfusion of blood. When leucodepletion was introduced, it was believed that there was only a theoretical risk that vCJD could be transmitted between humans via blood. Subsequent experiments in animals demonstrated that bovine spongiform encephalopathy (BSE) infectivity was transmissible through blood transfusion6 whilst, more recently, two definite cases and a further probable case of the transmission of vCJD through transfusion in humans have been reported.7,8 The emergence of new blood-borne infective agents and the consequent need for more elaborate pathogen inactivation and monitoring technologies will inevitably add to the marked increase in the cost of a unit of blood that has occurred in recent years.9 Exposure to donor blood can, of course, be reduced by using the patient's own (autologous) blood, pre-deposited or salvaged during operations. However, this approach has logistical problems, is costly and unsuitable for some patients, particularly those who are anaemic.10
Apart from the practical and physiological problems are the cultural and religious objections to receiving a blood transfusion. This is best illustrated by Jehovah's Witnesses and the biblical injunction in Leviticus (17:11): ‘Ye shall not eat the blood of no manner of flesh: for the life of all flesh is the blood thereof’. Jehovah's Witnesses interpret this edict so strictly that they refuse blood transfusions, even when their own or their child's life is threatened.
This paper gives an overview on the strategies employed to produce so-called blood substitute materials, focusing on the chemistry of the principal constituents and their clinical applications as alternatives to the transfusion of blood.
The need for blood substitutes
In principle, the use of so-called blood substitutes could solve many of the problems associated with using blood for transfusion. Specific clinical targets have been identified for such blood substitutes, most especially their use to replace acute blood loss during surgery or following trauma.11 In people who refuse blood transfusions or those unable to a receive transfusion for clinical reasons (e.g. patients with autoimmune haemolytic disease), an effective blood substitute could be life saving.There has been interest and significant research activity in the development and use of blood substitutes since the 1970s. Progress in this field was accelerated by the emergence of the human immunodeficiency virus (HIV) epidemic in the 1980s and the realisation that infectivity was transmissible via blood.12 The term ‘blood substitute’ when used in the present context is, however, inaccurate. Candidate materials should be more accurately referred to as ‘red cell substitutes’, ‘artificial oxygen carriers’, ‘anti-hypoxic drugs’ or, preferably, ‘oxygen therapeutics’. Put simply, they are fluids which, when injected into the blood, provide volume expansion and make a significant contribution to both systemic oxygen transport and tissue oxygenation.13 When coupled with the use of autologous blood, oxygen therapeutics will be a crucial part of future blood-conserving strategies to avoid patient exposure to donor blood and reduce the risk of contracting diseases transmissible via blood.
Strategies for developing blood substitute materials
Research and development on blood substitute materials has focused on two main avenues of investigation, which are:1. Fluids based on chemically-modified products of the naturally-occurring, oxygen-carrying respiratory pigment, haemoglobin (Hb), present in red blood cells (erythrocytes).
2. Emulsions of chemically inert, perfluorinated compounds called perfluorochemicals (PFCs) that dissolve oxygen and other gases.
There have thus emerged two distinct strategies for developing injectable oxygen carriers, namely the biomimetic and the abiotic approach, respectively.14 The former strategy mimics nature's way of delivering oxygen to tissues, whereas the latter approach uses totally synthetic chemicals to achieve the same result.
Haemoglobin
Chemistry and oxygen binding
Adult human Hb is a 64 kDa protein comprising four sub-units, each made up of a proteinaceous globin moiety covalently bound to a haem group comprising of a porphyrin derivative with an atom of iron in the oxidised state (Fe2+). Each of the four iron atoms in Hb can combine reversibly with one molecule of oxygen to give oxyhaemoglobin (HbO2). The globin portion of Hb consists of two pairs of polypeptides, the α chains containing 1441 amino acid residues and the β chains comprising 146 amino acids. The normal adult human Hb (HbA) molecule is therefore a tetramer, designated α2β2, which makes up ∼97–98% of the Hb in red blood cells. In the remaining 2–3% of Hb molecules (designated HbA2), the β chains are replaced with δ chains. The latter also have 146 amino acid residues, but 10 of them are different from those in their β counterparts. The globin sub-units of deoxygenated Hb are held together by electrostatic forces in a tight conformation which has a low affinity for oxygen. When oxygen binds to a haem group, these forces weaken, exposing the binding sites, resulting in a marked increase in the affinity of Hb for oxygen.Normal blood contains ∼145 g of Hb per litre and each gram of Hb binds to 1.39 mL of oxygen. A litre of blood can therefore carry approximately 200 mL of oxygen, compared to only ∼3 mL dissolved in simple solution in the plasma. Because of its iron content, Hb is a pigment that appears reddish when combined with oxygen and bluish when deoxygenated. Fully oxygenated arterial blood is therefore red in colour, whereas venous blood that has given up some of its oxygen to the respiring tissues has a bluish appearance.
The relationship between oxygen content of Hb and partial pressure of oxygen (pO2) is a characteristic sigmoid relationship, often called the oxygen dissociation curve (Fig. 1). The affinity of Hb for oxygen is affected by environmental conditions including pH, temperature and the concentration in the red blood cells of the anion, 2,3-diphosphoglycerate (2,3-DPG). 2,3-DPG reacts with Hb causing a decrease in its oxygen binding affinity with consequent oxygen offloading, thereby facilitating oxygenation of tissues. The oxygen affinity for Hb and blood is conventionally expressed by the P50 value, the partial pressure of oxygen when the blood is 50% saturated. A typical P50 for normal whole blood is ∼27 mmHg, whereas in the absence of 2,3-DPG it can fall to ∼15 mmHg. This explains why solutions of Hb, first tested as simple blood substitutes almost 100 years ago, were poor deliverers of oxygen to tissues. One solution to this problem was to covalently modify Hb with pyridoxal-5-phosphate (pyridoxylation), a natural coenzyme and functional analogue of 2,3-DPG, to reduce its oxygen affinity.14 This ensured that tissue oxygenation with the modified product was adequate.
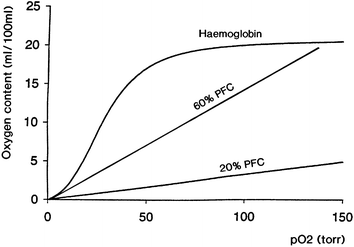 | ||
| Fig. 1 Schematic relationship between oxygen content and oxygen partial pressure (pO2) for haemoglobin (Hb) and a perfluorochemical (PFC) liquid. Note the linear relationship between these variables for the PFC. | ||
Hb as a blood substitute
Hb was an obvious choice for developing blood substitutes and the first intravascular infusions of Hb solution in humans were made by Wilson Sellards and the subsequent Harvard Nobel laureate George Minot at Massachusetts General Hospital, Boston, in 1916.15 They administered very small amounts of Hb to 33 subjects that were generally well tolerated. Subsequent infusions of human Hb produced mixed reactions in recipients and, in some cases, led to kidney toxicity. It is now known that such complications were caused primarily by residual fragments of the red cell membranes (stroma) and dimers produced from the dissociation of the tetrameric Hb molecules. Such Hb fragments were also rapidly excreted through the kidneys, typically within a few hours of being injected. This experience led to marked improvements in both the purification and stabilising processes used to prepare Hb for intravascular use.14 Chemical modifications used to stabilise Hb and improving its circulation time in the bloodstream include:1. Intramolecular cross-linking (e.g. with diaspirin) to form stable tetramers.
2. Conjugation (‘surface decoration’) with a polymer (e.g. polyethylene glycol).
3. Polymerisation (e.g. with glutaraldehyde or o-raffinose) producing intermolecularly cross-linked Hb.
4. Encapsulation of pyridoxylated Hb within a lipid membrane, producing so-called ‘synthetic red cells’.
Chemically-modified Hb is desirable as a blood substitute since the unmodified molecule is associated with unwanted cardiovascular effects, most notably a marked increase in blood pressure (hypertension) and a resultant fall in cardiac output.16,17 This occurs because Hb extravasated from the circulation binds avidly to the potent blood vessel dilator, nitric oxide (NO), derived from vascular endothelium, causing vasoconstriction and raising blood pressure.18 Other potential toxic effects of Hb which cause concern are the generation of toxic oxygen radicals that are well known to cause tissue damage in humans.16,17,19
Several chemically-modified Hbs have been developed commercially as oxygen-carrying blood substitutes and are in various stages of clinical evaluation (Table 1). Such materials include products based on human (HemAssist™, Baxter International, USA; Hemolink™, Hemosol Inc., Canada; PolyHeme™, Northfield, USA) and cow (bovine) blood (e.g. Hemopure™, Biopure Corporation, USA).15,17,20–26 One advantage of bovine Hb is that it is not necessary to adjust the oxygen affinity since this depends on chloride ions, rather than 2,3-DPG as in humans. PolyHeme™ contains polymerised human Hb and has been used to promote survival in patients with urgent blood loss who had refused a blood transfusion on religious grounds.27 The product is under evaluation in a major clinical trial as a life-sustaining pre-hospital (ambulance) therapy for severely-injured, mostly unconscious trauma patients (Table 1). This application takes advantage of a US Federal law that does not require patients to give informed consent when no alternative effective treatment (i.e. blood) exists.
| Product | Principal constituent | Country of origin | Clinical trials | Approved for clinical use | Year | Country | Approved indication(s) (planned in italics) |
|---|---|---|---|---|---|---|---|
| a Production ceased in 1994. b Severely injured bleeding trauma patients unable to provide informed consent for treatment. | |||||||
| Fluosol® | PFC | Japan | Yes | Yes | 1990 | USA, Europe | Coronary angioplastya |
| Perftoran® | PFC | Russia | Yes | Yes | 1996 | Russia | Blood avoidance in surgery |
| Trauma, ischaemia | |||||||
| Organ preservation | |||||||
| Hemopure™ | Bovine Hb | USA | Yes | Yes | 2001 | South Africa | Blood avoidance in surgery |
| Oxygent™ | PFC | USA | Yes | Pending | — | — | Blood avoidance in surgery |
| PolyHeme™ | Human Hb | USA | Yes | Pending | — | — | Pre-hospital trauma therapy b |
| Hemospan™ | Human Hb | USA | Yes | Pending | — | — | Blood avoidance in surgery |
| Oxycyte™ | PFC | USA | Yes | Pending | — | — | Blood avoidance in surgery |
| OxyVita™ | Bovine Hb | USA | No | — | — | — | Blood avoidance in surgery |
| Pher-O2™ | PFC | USA | No | — | — | — | Blood avoidance in surgery |
One newer product, Hemospan™ (Sangart, USA), contains human Hb conjugated to maleimide-treated polyethylene glycol (MalPEG).23,24,28 Briefly, the MalPEG-Hb was produced by the reaction of human Hb obtained from outdated blood with iminothiolane (Traut's reagent) to introduce sulfhydryl groups on selected lysine residues which were subsequently conjugated to the MalPEG. The resultant purified molecule has a molecular mass about twice that of Hemopure™23,24,28 This prevents extravasation from the circulation, thereby avoiding the hypertensive side effects with some products discussed already. Interestingly, Hemospan™ also has a higher affinity for oxygen (P50 = 5–6 mmHg) than blood and most other Hb-based blood substitutes. The rationale is to avoid excessive oxygenation of tissues and prevent compensatory arteriolar vasoconstriction that would contribute to hypertension. This has been confirmed in pre-clinical studies and clinical trials with Hemospan™ are in progress.23,24 This new paradigm shift in the design of blood substitutes is controversial and not without its critics. Nevertheless, a further new product, the bovine Hb-based OxyVita™ (OxyVita, USA), known previously as ‘zero-link’ Hb,29 has similar properties.
A landmark in blood substitute development was reached in 2001 when Hemopure™ received regulatory approval in South Africa for clinical use as an alternative to using blood during surgery in adults. Hemopure™ consists of bovine Hb that has been treated with glutaraldehyde (Table 2) which interacts with amino acid residues to cause intermolecular cross-linking between the tetramers. This yields a polymerised Hb molecule with an average molecular weight of 250 kD. The increase in size prevents the rapid extravasation of the Hb polymer, prolonging its dwell time in the blood. The manufacturer's state that the Hb for Hemopure™ is sourced from blood from cattle with a known pedigree and raised in non-BSE regions of the world. The non-human origin of the Hb in Hemopure™ means that it is likely to be accepted by some Jehovah's Witnesses. It is noteworthy that this blood substitute was first approved in a region of the world where a significant proportion of the adult population are infected with HIV and there are serious risks of further spreading the disease through transfusion of blood. In Western Europe and the USA, however, a bovine Hb-based product may be less acceptable to patients, primarily because of concerns about the transmission of BSE discussed already. There is also a significant risk that such animal Hb may adversely affect the human immune system, especially on repeat infusion, but this has not been studied systematically. Nevertheless, from a cost–benefit consideration, bovine Hb does represent a relatively inexpensive and abundant source of material for the commercial production of oxygen therapeutics. It has been estimated that up to 40 units of product (1 unit = ∼50 g Hb or 450 mL whole blood) could be obtained from a single cow.30
| Characteristic | Value |
|---|---|
| a Glutaraldehyde-polymerised bovine Hb (HBOC-201). b Denotes the partial pressure of arterial oxygen at which 50% of the Hb is saturated under standard conditions. | |
| Total Hb content/g dL−1a | 13.0 |
| Methaemoglobin content (%) | <5.0 |
| P 50/mmHg at 37 °Cb | 38.0 |
| pH | 7.6–7.9 |
| Colloid osmotic pressure/mmHg | 25.0 |
| Osmolarity/mOsm kg−1 | ∼300 |
| Viscosity/cP | 1.3 |
| Circulation half-time/h | ∼24 |
| Stability | >2 years at 2–37 °C |
Hb from microorganisms and plants
Human Hb has been produced from genetically modified micro-organisms31–34 and plants.35 This approach avoids using human or animal blood as starting material, thereby overcoming cultural and ethical objections to animal or human donor blood-derived Hb and the problem of transmitting infections. In principle, such recombinant molecules are virus free and do not contain residual red cell membrane contaminants. Plants are increasingly being targeted as inexpensive and convenient systems for the large-scale production of valuable recombinant proteins.36,37 This approach to protein generation is often called ‘molecular farming’. Tobacco (Nicotiana tabacum) has, for example, been used successfully as an expression system for human therapeutic proteins, including erythropoietin and growth hormone.38 Plants are an ethically acceptable system representing perhaps an obvious choice for the production of human Hb on a commercial scale. It remains to be seen, however, whether a fully functional human Hb can be expressed and extracted intact from plants, but it is a challenge worth pursuing.The most advanced rHb material was called Optro™ (Baxter, USA) which was produced in Escherichia coli and remains, to date, the only recombinant product to enter clinical trials as an oxygen carrier. Optro™ contained rHb1.1, a Hb mutant carrying a mutation at position 108 on the β globin chain where lysine was substituted for asparagine31,32 The resultant molecule was thus similar to the naturally-occurring Presbyterian Hb mutant first reported in 1978.39 Importantly, this mutation conferred on rHb1.1 a low oxygen binding affinity in the absence of 2,3-DPG making it a candidate for evaluation as an injectable oxygen therapeutic. A stable tetrameric structure of the rHb1.1 was achieved by the genetic fusion of the two α chains via a glycine.31 This eliminated renal toxicity and improved circulation time in the blood. Optro™ was evaluated in both pre-clinical and clinical trials and, although the material was generally well tolerated, the programme was terminated following reports of fever, gastrointestinal side effects and hypertension in volunteers.33,34,40 The latter two effects of Optro™ were attributed mainly to NO scavenging by rHb1.1.
Recombinant Hbs are a logical way forward for oxygen therapeutics since ‘tailor-made’ molecules can, in principle, be produced. Future work should take account of the discovery that NO can bind not only to iron in haem in Hb, but also to reactive sulfhydryl groups of cysteine residues in the molecule.41 When Hb delivers oxygen to tissues, it also releases NO from its cysteine residues and this, in turn, causes vasodilation of capillaries to facilitate oxygenation. Recombinant technology has been used to produce Hb mutants with altered affinity for NO, thereby overcoming the transient, but unwanted, hypertensive (raised blood pressure) side effects that are one disadvantage of Hb-based blood substitutes.42–44
In 2004, a multi-centre European team, led by the University of Nottingham, initiated a new project, entitled Genomics and Blood Substitutes for 21st Century Europe (‘EuroBloodSubstitutes’), funded by the European Union, to develop a technological baseline for producing novel haem proteins and blood substitute components using micro-organisms as ‘cell factories’.45 Bacteria (E. coli) and yeasts (Pichea pastoris, Saccharomyces cerevisiae) are being used in the project because such organisms are already being exploited for the commercial production of human pharmaceuticals (e.g. insulin, growth hormone) and food additives (e.g. enzymes), typically under large-scale, bioreactor conditions.46–50 The challenges faced by this new project, however, are to genetically engineer the micro-organisms to synthesise haem proteins, extract and purify them without adversely altering their biochemical characteristics, and scale-up the production of Hb to a commercial scale. Even a predicted annual use of a unit dose of ∼50 g Hb in, say, 5% of the European population would require the production of ∼2000 kg of protein, in itself a major commercial undertaking.
Invertebrate Hbs
One exciting development has been the extraction of Hb from the earthworm (Lumbricus terrestris) and the common marine worm (Arenicola marina) for testing as possible blood substitutes. Their Hbs are naturally-occurring polymers up to 50 times larger than human Hb. Earthworm Hb, for example, is not toxic in rats and mice and has a normal oxygen-carrying capacity.51 In addition to gaining new insights into the properties and behaviour of such polymeric Hbs, perhaps as models for the design of new materials for blood substitutes, one challenge will be to obtain sufficient material, in a highly purified form, for further crucial safety (especially immunogenicity) and efficacy tests.Artificial red blood cells
On-going research has focused on the encapsulation of Hb within biodegradable polymer membranes as the first step to producing so-called ‘artificial red cells’. Other refinements have been to entrap anti-oxidant enzymes, especially superoxide dismutase and catalase, with Hb to create a microenvironment that much more closely resembles that of the natural red blood cell.52 This is ambitious, yet exciting, research that will soon yield a range of advanced Hb-based blood substitutes for pre-clinical and clinical testing. Such materials also have promise as novel drug delivery systems and therapeutic agents, as reviewed by Chang.53Synthetic, fluorinated compounds
Chemical properties
The second, abiotic approach to the development of blood substitutes involves the use of synthetic PFC liquids that dissolve large volumes of oxygen.13,14 PFCs are linear, cyclic or polycyclic hydrocarbons in which hydrogen atoms have been substituted with fluorine. The two compounds most widely used in biological systems are perfluorodecalin (C10F18), a bicyclic perfluorinated alkane, and bromoperfluoro-n-octane (empirical formula: C8F17Br, known by the generic name of perflubron), a linear molecule with a terminal bromine atom (Fig. 2).13,14,54,55 Liquid PFCs are colourless, odourless and have specific gravities about twice that of water. PFCs were first produced commercially during World War II as part of the Manhattan Project, in the search for inert handling materials that could resist corrosion by the highly reactive uranium isotopes being synthesised for the first atomic bomb.13 PFCs are extremely inert owing to the high strength of the carbon-fluorine bond (∼480 kJ mol−1) and the protective effects that the large, electron-rich fluorine atoms confer on the underlying carbon backbone shielding it from chemical or enzymatic attack.13,14,55 Commercial applications of PFCs include their use as industrial lubricants, laser coolants and anti-corrosion agents. Teflon® or poly(tetrafluoroethylene), the solid protective anti-stick coating on household cookware and frying pans, is a polymerised and highly corrosion resistant PFC. The inertness of PFCs also makes them unreactive in the body. The molecules are sequestered by phagocytic cells of the monocyte/macrophage lineage (formerly known as the reticuloendothelial system). They subsequently diffuse back into the blood where they are carried in plasma lipids to the lungs and exhaled intact as a vapour.13,14,54,55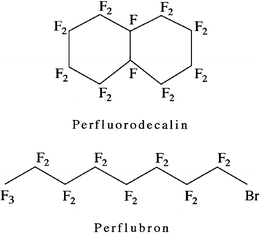 | ||
| Fig. 2 Chemical structures of two PFCs, perfluorodecalin (top) and perflubron (bottom). | ||
Gas solubility
PFCs have the highest gas-dissolving capacities of any liquids. The solubility of respiratory gases, for example, is related to the molecular volume of the dissolving gas and decreases in the order CO2 ≫ O2 > N2. The solubility of oxygen in PFC liquids (37 °C, 1 atm) used for biomedical applications is ∼40–50 vol.%, as compared to 2.5 vol.% for water; carbon dioxide solubility in the same liquids can be >200 vol.%.13,14,54,55 Unlike the active binding of oxygen to the haem sites of Hb, oxygen dissolution in PFCs is a passive process, in which gas molecules occupy ‘cavities’ within the PFC liquid. Consequently, in contrast with the sigmoid binding curve of oxygen to Hb, the solubility of the gas in a PFC liquid at a given temperature is directly proportional to the pO2, essentially obeying Henry's Law (Fig. 1).Biomedical applications: emulsification for intravascular use
PFC liquids were first studied as biologic gas carriers in the 1960s.13,54 Interest in the potential biomedical uses of such compounds was provoked by the pioneering experiments of Clark which demonstrated that mice could survive whilst ‘breathing’ in an oxygenated fluorocarbon liquid (FC-80, butyltetrahydrofuran) in which they were immersed (Fig. 3).56 PFC liquids have, for example, been exploited in medical biotechnology for the liquid ventilation of premature babies with respiratory disease57 and increasing gas supply to animal, plant and microbial cells in vitro.58,59 For use as injectable oxygen carriers, however, PFCs must be prepared as emulsions with an average target droplet diameter of 0.1–0.2 µm or less.13,14,55 This is because PFC liquids are immiscible in aqueous systems, including the blood plasma. The first demonstration that a fluorocarbon emulsion could be used to replace the blood of laboratory animals was reported by Geyer and colleagues in 1968.60 Since then, a sizeable literature describing aspects of the formulation, preparation, stabilisation and characterisation of such emulsions, together with some of the biomedical properties of the final product(s) and their individual constituents, has emerged,13,14,54 but the details are beyond the scope of the present review.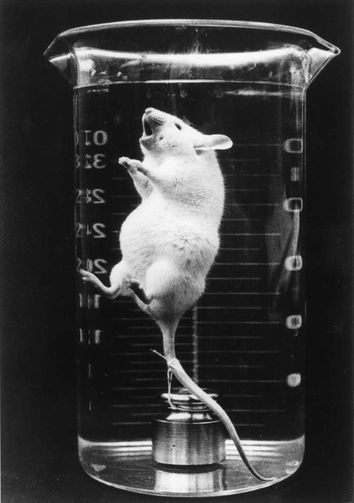 | ||
| Fig. 3 Mouse ‘breathing’ when totally immersed in an oxygen saturated perfluorocarbon (FC-80, butyltetrahydrofuran) liquid (photograph courtesy Professor Leland Clark). This pioneering experiment stimulated great interest in the development of fluorocarbons as respiratory gas carriers in biological systems. | ||
| Characteristic | Fluosol® | Perftoran® |
|---|---|---|
| a Refers to preferred conditions for storage of ‘stem’ emulsion containing PFCs. | ||
| PFCs | Perfluorodecalin | Perfluorodecalin |
| Perfluorotripropylamine | Perfluoromethylcyclopiperidine | |
| Emulsifiers (surfactants) | Pluronic F-68 | Proxanol 268 |
| Potassium oleate | Egg yolk phospholipids | |
| Total PFC content (% w/v) | 20.0 | 20.0 |
| Average droplet diameter/µm | <0.2 | 0.07 |
| pH | 7.3 | 7.3 |
| Osmolarity/mOsm kg−1 | 410 | 300 |
| Viscosity/cP | 2.8 | 2.3 |
| Optimum storage conditionsa | Frozen | Frozen |
| Characteristic | Value |
|---|---|
| PFC content (% w/v) | 60.20 ± 0.06 |
| Mean droplet diameter/µm | 0.17 ± 0.01 |
| pH | 7.10 ± 0.04 |
| Viscosity/cP | 4.0 ± 0.1 |
| Osmolarity/mOsm kg−1 | 304 ± 6 |
| Oxygen solubility (vol%) | 19.3 |
| Carbon dioxide solubility (vol%) | 157.0 |
| Stability | >1 year at 5–10 °C |
Oxygent™ has been tested in over 1300 patients in large, multi-centre clinical trials. In one study, infusion of Perflubron emulsion into patients who simultaneously breathed supplementary oxygen was more effective than autologous blood or colloid solution in maintaining cardiovascular function and tissue oxygen supply.65 In a separate trial with 492 general surgery patients, treatment with Oxygent™ reduced both the frequency and volume of blood used during surgery.66 In some trials, Oxygent™ was used in conjunction with a procedure called acute normovolaemic haemodilution (ANH), a technique designed to avoid the need for allogeneic blood transfusions in moderate to high blood loss surgical procedures (Table 1). Immediately prior to surgery, several units of blood were removed from the patient and Oxygent™ used to maintain tissue oxygenation. Following surgery, the removed blood was re-infused. In 2001, Alliance terminated a Phase III cardiac surgery study with Oxygent™ to evaluate an unexpected increased incidence of stroke in some patients that was subsequently ascribed by the company as being due to overly aggressive pre-surgical ANH that had been part of the study protocol.67 For many, this was an unexpected setback for a product that was widely expected to be granted regulatory approval for routine clinical use. Nevertheless, in 2005, new licensing, development and marketing agreements with pharmaceutical companies in China, Europe and Canada were announced for the further clinical and regulatory development of the drug for organ protection against hypoxia during major surgery.
Other fluorocarbon products
As part of the on-going evaluation of the potential of PFC-based materials as injectable oxygen carriers, other ‘second-generation’ products are in commercial development (Table 1). These include Oxycyte™ (Synthetic Blood International, USA) and Pher-O2 (Sanguine Corporation, USA). A Phase I (safety) clinical trial with Oxycyte™ was completed in 2004 with satisfactory outcomes and Phase II clinical trials with for the prevention of tissue hypoxia during orthopaedic and cardiac surgery are planned.Engagement with stakeholders
Whilst the physiological and economic aspects of risk in transfusion have been well studied, surprisingly little attention has focused on the perceptions of benefit and risk associated with the transfusion of blood, blood products or, most especially, potential blood substitutes.5,68 It is becoming recognised that public attitudes will play a crucial role in realising the potential of scientific and technological advances.69 It is timely to ask:1. What relevant stakeholder groups know about transfusion.
2. How safe people perceive blood and blood products to be.
3. How the latter information might influence their own or others' perceptions of risk linked to transfusion.
4. To what extent will approved blood substitutes be preferred over a person's own or donor blood.
In two recent investigations, Ferguson et al.70 and Fleming et al.71 studied the perceptions of risk, effectiveness and ethicality of different hypothetical transfusion options among young adults in the UK. Overall, the results showed that donor blood was considered significantly more effective and more ethical than all the potential blood substitutes evaluated. Blood substitutes based on synthetic ‘chemicals’ (i.e. abiotic materials) were rated by respondents as similar to recombinant Hb-based substitutes in terms of effectiveness, but were viewed as more ethical. Substitutes based on bovine Hb were perceived as the least ethical. All the blood substitutes were perceived as having similar effectiveness, albeit to a lower extent than for donor blood. Such information on what stakeholders perceive to be the benefits and risks of the receipt of blood and blood substitutes will inform future transfusion strategies and the design of new decision aids and physician training aids, especially on blood substitutes as the latter become approved for routine clinical use.
Concluding remarks
To date, progress in the development of blood substitutes has focused on the exploitation of Hb from human or animal blood, or the use of emulsified PFCs. A bovine Hb product has been approved for clinical use, albeit in a region of the world where blood-borne disease is currently endemic. For animal Hbs, issues of immunoreactivity, disease transmission and patient acceptance are still unresolved. Recombinant technology together with microbial and plant ‘cell factories’ are being used to produce novel haem proteins and blood substitute components, but the challenge will be to produce highly purified material on a commercial scale. Polymeric invertebrate Hbs are also being targeted as model haem proteins or sources of raw materials for future blood substitutes. The advantage of the PFC-based approach to blood substitutes is that the constituents are synthetic and problems of source and purity are less problematic. PFCs have multi-faceted applications in medicine and biotechnology, with most interest currently focused on their use as temporary tissue oxygenation fluids during surgery or therapy. Feedback from lay and professional stakeholders is increasingly being sought and used to inform future transfusion strategies and the design of new information materials. The future therapeutic use of the biomimetic and abiotic blood substitutes outlined in this paper will increase transfusion options for patients and practitioners and contribute to modern blood conserving strategies for the 21st century.References
- B. D. Spiess, R. B. Counts and S. A. Gould, Perioperative Transfusion Medicine, Williams & Wilkins, Baltimore, 1998, p. 3 Search PubMed.
- M. L. Finucane, P. Slovic and C. K. Mertz, Transfusion, 2000, 40, 1017–1022 CrossRef CAS.
- K. C. Lowe, K. Farrell, E. M. P. Ferguson and V. James, Artif. Cells, Blood Substitutes, Immobilization Biotechnol., 2001, 29, 179–189 CrossRef CAS.
- E. M. P. Ferguson, K. Farrell, K. C. Lowe and V. James, Transfus. Med., 2001, 11, 129–135 CrossRef CAS.
- K. C. Lowe and E. Ferguson, J. Intern. Med., 2003, 253, 498–507 CrossRef CAS.
- N. Hunter, J. Foster, A. Chong, S. McCutcheon, D. Parnham, S. Eaton, C. MacKenzie and F. Houston, J. Gen. Virol., 2002, 83, 2897–2905 Search PubMed.
- C. A. Llewelyn, P. E. Hewitt, R. S. G. Knight, K. Amar, S. Cousens, J. Mackenzie and R. G. Will, Lancet, 2004, 363, 417–421 CrossRef CAS.
- A. H. Peden, M. W. Head, D. L. Ritchie, J. E. Bell and J. W. Ironside, Lancet, 2004, 364, 527–529 CrossRef.
- M. Amin, D. Fergusson, A. Aziz, K. Wilson, D. Coyle and P. Héber, Transfus. Med., 2003, 13, 275–285 CrossRef CAS.
- J. Etchason, L. Petz, E. Keeler, L. Calhoun, S. Kleinman, C. Snider, A. Fink and R. Brook, New Engl. J. Med., 1995, 332, 719–724 CrossRef CAS.
- J. C. Fratantoni, in Blood Cell Substitutes, ed. A. S. Rudolph, R. Rabinovici and G. Z. Feuerstein, Marcel Dekker, New York, 1998, p. 29 Search PubMed.
- K. A. Sepkowitz, New Engl. J. Med., 2001, 344, 1764–1772 CrossRef CAS.
- K. C. Lowe, Tissue Eng., 2003, 9, 389–399 CrossRef CAS.
- J. G. Riess, Chem. Rev., 2001, 101, 2797–2919 CrossRef CAS.
- E. E. Moore, J. Am. Coll. Surg., 2003, 196, 1–17 CrossRef.
- A. I. Alayash, Nat. Rev. Drug Discovery, 2004, 3, 152–159 CrossRef CAS.
- P. W. Buehler and A. I. Alayash, Transfusion, 2004, 44, 1516–1530 CrossRef CAS.
- J. P. Wallis, Transfus. Med., 2005, 15, 1–11 CrossRef CAS.
- L.-H. Yeh and A. I. Alayash, J. Intern. Med., 2003, 253, 518–526 CrossRef CAS.
- C. P. Stowell, J. Levin, B. D. Spiess and R. M. Winslow, Transfusion, 2001, 41, 287–299 CrossRef CAS.
- T. M. S. Chang, J. Intern. Med., 2003, 253, 527–535 CrossRef CAS.
- T. J. Reid, Transfusion, 2003, 43, 280–287 CrossRef CAS.
- R. M. Winslow, J. Intern. Med., 2003, 253, 508–517 CrossRef CAS.
- R. M. Winslow, Artif. Cells, Blood Substitutes, Biotechnol., 2005, 33, 1–12 Search PubMed.
- Blood Substitutes, ed. R. M. Winslow, Academic Press, London, 2005 Search PubMed.
- H. W. Kim and A. G. Greenburg, Artif. Organs, 2004, 28, 813–828 CrossRef CAS.
- S. A. Gould, E. E. Moore, D. B. Hoyt, P. M. Ness, E. J. Norris, J. L. Carson, G. A. Hides, I. H. G. Freeman, R. DeWoskin and G. S. Moss, J. Am. Coll. Surg., 2002, 195, 445–455 CrossRef.
- K. D. Vandegriff, A. Malavalli, J. Woodridge, J. Lohman and R. M. Winslow, Transfusion, 2003, 43, 509–516 CrossRef CAS.
- B. Matheson, H. E. Kwansa, E. Bucci, A. Rebel and R. C. Koehler, J. Appl. Physiol., 2002, 93, 1479–1486 Search PubMed.
- S. M. Cohn, New Horiz., 1999, 7, 54–60 Search PubMed.
- D. Looker, D. Abbott-Brown, P. Cozart, S. Durfee, S. Hoffman, A. J. Mathews, J. Miller-Roehrich, S. Shoemaker, S. Trimble, G. Fermi, N. H. Komiyama, K. Nagai and G. L. Stetler, Nature, 1992, 356, 258–260 CrossRef CAS.
- D. Looker, A. J. Mathews, J. O. Neway and G. L. Stetler, Methods Enzymol., 1994, 231, 364–374 CAS.
- M. L. Gerber, G. L. Stetler and D. Templeton, in Artificial Red Cells, ed. E. Tsuchida, Wiley, Chichester, 1995, p. 187 Search PubMed.
- S. H. Zuckerman, M. P. Doyle, R. Gorczynski and G. J. Rosenthal, Artif. Cells, Blood Substitutes, Immobilization Biotechnol., 1998, 26, 231–257 CAS.
- W. Dieryck, J. Pagnier, C. Poyart, M. C. Marden, V. Gruber, P. Bournat, S. Baudino and B. Mérot, Nature, 1997, 386, 29–30 CrossRef CAS.
- S. Schillberg, R. Fischer and N. Emans, Cell. Mol. Life Sci., 2003, 60, 433–445 CrossRef CAS.
- R. M. Twyman, E. Stiger, S. Schillberg, P. Cristou and R. Fischer, Trends Biotechnol., 2003, 21, 570–578 CrossRef CAS.
- J. W. Larrick and D. W. Thomas, Curr. Opin. Biotechnol., 2001, 12, 411–418 CrossRef CAS.
- W. F. Moo-Pen, J. A. Wolff, G. Simon, M. Vacek, D. L. Due and M. H. Johnson, FEBS Lett., 1978, 92, 53–56 CrossRef.
- M. K. Viele, R. B. Weiskopf and D. Fisher, Anesthesiology, 1997, 86, 848–858 CrossRef CAS.
- L. Jia, C. Bonaventura, J. Bonaventura and J. S. Stamler, Nature, 1996, 380, 221–226 CrossRef CAS.
- D. H. Doherty, M. P. Doyle, S. R. Curry, R. J. Vali, T. J. Fattor, J. S. Olsen and D. D. Lemon, Nat. Biotechnol., 1998, 16, 672–676 CrossRef CAS.
- J. S. Olson, E. W. Foley, C. Roggie, A.-L. Tsai, M. P. Doyle and D. D. Lemon, Free Radical Biol. Med., 2004, 36, 685–697 CrossRef CAS.
- D. H. Maillett and J. S. Olsen, in Blood Substitutes, ed. R. W. Winslow, Academic Press, London, 2005, p. 354 Search PubMed.
- K. C. Lowe, Artif. Cells, Blood Substitutes, Biotechnol., 2006 Search PubMed , in press.
- D. B. Archer, Curr. Opin. Biotechnol., 2000, 11, 478–483 CrossRef CAS.
- J. M. Cregg, J. L. Cereghino, J. Shi and J. R. Higgins, Mol. Biotechnol., 2000, 16, 23–52 Search PubMed.
- J. R. Swartz, Curr. Opin. Biotechnol., 2001, 12, 195–201 CrossRef CAS.
- G. P. L. Cereghino, J. L. Cereghino, C. Iigen and J. M. Cregg, Curr. Opin. Biotechnol., 2002, 13, 329–332 CrossRef.
- D. B. Archer and P. S. Dyer, Curr. Opin. Microbiol., 2004, 7, 499–504 CrossRef CAS.
- R. E. Hirsch, L. A. Jelicks, B. A. Wittenburg, D. K. Kaul, H. L. Shear and J. P. Harrington, Artif. Cells, Blood Substitutes, Immobilization Biotechnol., 1997, 25, 429–444 CAS.
- T. M. S. Chang, in Principles of Tissue Engineering, 2nd edn, ed. R. P. Lanza, R. Langer and J. Vacanti, Academic Press, San Diego, 2000, p. 601 Search PubMed.
- T. M. S. Chang, Nat. Rev. Drug Discovery, 2005, 4, 221–235 CrossRef CAS.
- K. C. Lowe, Blood Rev., 1999, 13, 171–184 CrossRef CAS.
- J. G. Riess, Artif. Cells, Blood Substitutes, Biotechnol., 2005, 33, 47–63 Search PubMed.
- L. C. Clark and F. Gollan, Science, 1966, 152, 1755–1756 CAS.
- J. S. Greenspan, M. R. Wolfson and T. H. Shaffer, Semin. Perinatol., 2000, 24, 396–405 Search PubMed.
- K. C. Lowe, M. R. Davey and J. B. Power, Trends Biotechnol., 1998, 16, 272–277 CrossRef CAS.
- K. C. Lowe, J. Fluorine Chem., 2002, 118, 19–26 CrossRef CAS.
- R. P. Geyer, R. G. Monroe and K. Taylor, in Organ Perfusion and Preservation, ed. J. C. Norman, J. Folkman, W. G. Hardison, L. E. Rudolf and F. J. Veith, Appleton Century Crofts, New York, 1968, p. 85 Search PubMed.
- K. C. Lowe, in Blood Substitutes, ed. R. W. Winslow, Academic Press, London, 2005, p. 276 Search PubMed.
- E. Maevsky, G. Ivanitsky, L. Bogdanova, O. Axenova, N. Karmen, E. Zhiburt, R. Senina, S. Pushkin, I. Maslennikov, A. Orlov and I. Marinicheva, Artif. Cells, Blood Substitutes, Biotechnol., 2005, 33, 37–46 Search PubMed.
- E. Maevsky, G. Ivanitsky, B. I. Islamov, V. V. Moroz, L. A. Bogdanova, N. B. Karmen, S. Y. Pushkin and I. A. Maslennikov, in Blood Substitutes, ed. R. W. Winslow, Academic Press, London, 2005, p. 288 Search PubMed.
- P. E. Keipert, in Blood Substitutes, ed. R. W. Winslow, Academic Press, London, 2005, p. 312 Search PubMed.
- D. R. Spahn, R. van Brempt, G. Theilmeier, J.-P. Reibold, M. Welte, H. Heinzerling, K. M. Birck, P. E. Keipert and K. Messmer, Anesthesiology, 1999, 91, 1195–1208 CrossRef CAS.
- R. J. Frumento, L. Mongero, Y. Naka and E. Bennett-Guerrero, Anesth. Analg., 2002, 94, 809–814 Search PubMed.
- E. Niiler, Nat. Biotechnol., 2002, 20, 962–963 CrossRef CAS.
- E. Ferguson, P. Fleming, E. Townsend and K. C. Lowe, in New Developments in Blood Transfusion Research, ed. F. Columus, Nova, Hauppauge, 2006, in press Search PubMed.
- C. Wallace, Biologist, 2005, 52, 263–264 Search PubMed.
- E. Ferguson, J. Leaviss, E. Townsend, P. Fleming and K. C. Lowe, Transfus. Med., 2005, 15, 401–412 CrossRef CAS.
- P. Fleming, E. Ferguson, E. Townsend and K. C. Lowe, Artif. Cells, Blood Substitutes, Biotechnol., 2006 Search PubMed , in press.
| This journal is © The Royal Society of Chemistry 2006 |