Textural and electrochemical properties of carbon replica obtained from styryl organo-modified layered double hydroxide†
Fabrice
Leroux
*a,
Encarnacion
Raymundo-Piñero
b,
Jean-Marie
Nedelec
a and
François
Béguin
b
aLaboratoire des Matériaux Inorganiques, UMR 6002-CNRS, Université Blaise Pascal, 24 av. des Landais, 63177 Aubière cedex, France. E-mail: fleroux@chimtp.univ-bpclermont.fr
bCentre de Recherche sur la Matière Divisée, 1B rue de la Férollerie, 45071 Orléans, cedex2, France
First published on 16th March 2006
Abstract
Negative carbon replicas were obtained after pyrolysis of organo-modified layered double hydroxides (LDH) and dissolution of the inorganic moiety. The hybrid organic–inorganic precursors consisted of polystyrene sulfonate incorporated directly between LDH sheets of cationic composition Zn2Al or after in situ polymerisation of the monomers. The preparation conditions of the hybrid phase and the constraint imparted by the inorganic framework were found to affect strongly the textural properties. The carbons sequestered into the LDH intersheet gap present a textural hierarchy, i.e. a pronounced microporous character (micropore volume up to 1.07 mL g−1 and SBET = 2300 m2 g−1) and a mesoporosity. The nanotexture of the carbon replica was characterized by Raman spectroscopy. Finally, the carbons tested by cyclic voltammetry and complex impedance spectroscopy in two-electrode electrochemical supercapacitors in sulfuric acid medium present a capacitive behavior, i.e. a quasi rectangular response of intensity versus potential and a phase angle in the Z″–Z′ impedance plane close to π/2.
1. Introduction
Under specific conditions, some organic molecules once confined into an inorganic host structure give rise after pyrolysis to carbonaceous materials with interesting nanotextural properties. This so-called template carbonisation has a strong impact on the particle size, shape and porosity of the resulting carbons.1 It was found that the gas and water release during the decomposition of the inorganic framework are the main factors which influence the adsorption properties of the final carbon. The presence of the inorganic framework during the calcination is found to hinder the cross-linking reactions able to occur and therefore the carbonaceous materials develop large microporosity. Illustrated at first by 2D-host structures such as montmorillonite and using either polymer2,3 or other carbon sources,4–6 this concept was recently extended to 3D type frameworks such as mesoporous silica7–11 or zeolites.12As far as template carbonization is concerned, previous works have also involved hydrotalcite-type materials,13–16 so-called layered double hydroxides (LDHs). LDHs are described with the ideal formula, [MII1−xMxIII(OH)2]x+intra[Am−x/m·nH2O]inter, where MII and MIII are metal cations, A the anionic counterpart, and intra and inter denote the intralayer and interlayer domains, respectively. The structure consists of brucite-like layers constituted of edge-sharing M(OH)6 octahedra. Partial MII to MIII substitution induces a positive charge to the layers, balanced by the presence of the interlayered anions. In ref. 13, the authors observed that, for a given organic precursor, the carbons present similar specific surface area regardless of the nature of the LDH matrices. Using poly(styrene) sulfonate (PSS) as the carbon source, the obtained specific surface area measured by nitrogen adsorption was 1025 m2 g−1.
Our previous papers dealing with the PSS–LDH hybrid phases have shown that, apart from the direct PSS incorporation, interleaved styrene sulfonate monomers may connect each other and form polymer chains between the inorganic slabs.17–19 Taking into account that the preparation conditions, i.e. polymer entrapping versusin situ polymerisation, are different, we surmise that the porous texture of the carbon replica obtained after pyrolysis of the hybrids may be affected. This concept was partially enlightened in a feature article dedicated to inorganic organic LDH assembly.20 Therefore in this paper, several methods, t-plot, αs and Dubinin–Astakhov, were used to assess the nanotextural characteristics. The structure of the resulting carbon materials was characterized by Raman spectroscopy. This non-destructive technique gives information on the medium range order.21,22
In addition to nitrogen gas adsorption, the electrochemical adsorption behavior of the resulting carbons was also characterized, using them as electrodes of electrical double layer capacitors (EDLCs), often called supercapacitors. Indeed, it is widely accepted that a well balanced micro-, mesoporosity of carbons is very profitable for this application.23 Since the main application of supercapacitors is for short pulses or high power delivery, a reasonable proportion of mesopores is required to favor a quick diffusion of ions. The ions themselves are trapped in the micropores, therefore a well-developed surface area is needed. Finally, our previous work on carbons prepared in MCM-48 or SBA-15 3D-replica have demonstrated that a regular interconnection of micropores and mesopores is extremely advantageous to accelerate the whole process of ion transfer from the solution to the active carbon surface.24–26 By contrast, with an activated carbon, these two kinds of pores are randomly distributed. Hence, there is an interest to study the template carbons from LDHs hybrids for supercapacitors, because we may expect that microporous regions will be separated by 2D (or slit-shape) mesopores liberated by the removal of the inorganic framework. In this paper, the capacitive behavior of the carbon replica is tested in sulfuric acid medium by means of potentiostatic and galvanostatic techniques as well as by complex impedance spectroscopy.
2. Experimental section
2.1. Chemicals
Sodium vinylbenzene-4-sulfonate (VBS, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–C6H4–SO3−Na+ (Fluka, >90%)), syndiotactic poly(styrene sulfonate) (PSS, Acros, Mw = 700
CH–C6H4–SO3−Na+ (Fluka, >90%)), syndiotactic poly(styrene sulfonate) (PSS, Acros, Mw = 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), ZnCl2·5H2O (Acros), Al(NO3)3·9H2O (Acros, 99%), NaOH (Acros, 97%).
000 g mol−1), ZnCl2·5H2O (Acros), Al(NO3)3·9H2O (Acros, 99%), NaOH (Acros, 97%).
2.2. Preparation of the LDH hybrid materials
The so-called organized assembly method was used for the preparation of the hybrid materials. In all cases, the Zn∶Al LDH host structure was the same, with a Zn to Al cation ratio equal to 2. Typically, the organic molecular ions are dissolved in decarbonated water in amount corresponding to five times the anion exchange capacity of the solid, expected from a complete reaction between the initial Zn and Al inputs.18 Three different hybrid phases were prepared by direct incorporation of PSS or by incorporation of VBS followed or not by in situ polymerisation. The hybrid materials, PSS/Zn2Al and VBS/Zn2Al, were characterized in a previous paper.182.3. Carbonization of the organic part
Pyrolysis of the hybrid materials was performed in a tubular furnace under constant nitrogen gas flow (30 mL min−1), according to the procedures used by Putyera et al.13 The samples were treated at 600 °C for 3 h (heating rate of 10 °C min−1), then cooled down at room temperature. The powder was vigorously stirred in 1 mol L−1 HCl for 72 days in order to dissolve the inorganic moiety The obtained black powders were then washed several times with decarbonated water.2.4. Analysis of the porous texture by nitrogen adsorption
A Coulter apparatus was used for the nitrogen adsorption/desorption measurements at 77 K. The samples were outgassed at 150 °C under vacuum for 12 h. The specific surface area was calculated from the BET, Langmuir and Kaganer methods. The total pore volume was calculated taking the volume of nitrogen adsorbed at P/P0 = 0.99.The micropores volumes were determined by applying t-plot,27,28αs and Dubinin–Astakhov (D–A)29–33 methods. For t-plot and αs methods, the non-porous carbon Super S® (an acetylene black from Alpha Chemicals) was taken as reference.
The mesopore size was calculated taking the Barrett–Joyner–Halenda (BJH) traditional method using the desorption data.
2.5. Raman study
Raman spectra were recorded in the back scattering geometry on a Jobin-Yvon T64000 spectrometer equipped with a liquid nitrogen cooled CCD detector. The excitation beam is provided by the 514.5 nm line of an argon ion laser (Spectra physics 2017). The laser beam (about 1 µm2) is focused on the sample through an Olympus confocal microscope with ×100 magnification. The measured power at the sample level was kept low (less than 15 mW) in order to avoid any damage of the material. Motorized three axes stage and direct imaging allowed to select samples study zones, and several spectra have been recorded for each sample. The Raman scattered light was collected with the microscope objective at 180° from the excitation and filtered with an holographic Notch filter before being dispersed by a 1800 grooves mm−1 diffraction grating. The spectral resolution was estimated to be about 2 cm−1. All spectra were recorded at room temperature. Raman spectra of commercial graphite and Super S® were recorded as carbon references. The spectra have been corrected from the baseline, and deconvoluted using Lorentzian lines in the 800–2000 cm−1 spectral region.2.6 Electrochemical measurements
The carbon materials were tested in symmetric two-electrode capacitors using a teflon Swagelok® cell. The electrodes were pressed from a mixture of active material (80%), Super S® (15%), and polyvinylidene fluoride (PVDF, Kynar Flex 2801, Atochem, 5%). The positive and negative electrodes of comparable mass, ranging from 7 to 12 mg, are electrically isolated by a glassy fibrous separator. The electrolytic solution was 1 mol L−1 H2SO4 in water. The electrochemical characterizations were performed by voltammetry (scan rate of 2, 5 and 10 mV s−1) and galvanostatic cycling using a VMP (Biologic, France) multi-channel potentiostat/galvanostat. The galvanostatic current was set up equal to the potentiodynamic response under a rate of 2 mV s−1. For cyclic voltammetry, the capacitance was calculated from the recorded current value (I in mA) in the investigated potential domain divided by the sweep rate value (dV/dt in V s−1), and reported to the mass of the active material (g); C = I/(dV/dt) [(A/g)/(V/s) = F/g]. For the galvanostatic cycling, the slope of the potential versus time curve was used taking the time of discharge, td, over the voltage range V; C = I*td/(V/2). Impedance spectroscopy measurements were carried out in the frequency range from f = 10 Hz to 1 MHz using a VMP2 (Biologic, France). The capacitance is calculated using the following equation, C = 1/(2π*f*Z″), where Z″ is the imaginary impedance. The latter relation is valid for low frequency when the phase angle is close to π/2.In the following and for a sake of clarity, the carbon materials are noted C-VBS and C-PSS in absence of sequestration of the organic (macro)-molecular ion, and C-ZVBS and C-ZPSS from the hybrid organic inorganic materials. C-ZVBSpol denotes C-ZVBS obtained after a first thermal treatment under air to provocate the in situ polymerisation of interleaved VBS.
3. Results and discussion
3.1. Textural properties
Puteyra et al. have shown that, for the same organic anion, the carbon content related to the LDH layer charge was not the key factor in obtaining large surface area.13 However, in our case, a hydrotalcite-like material of high layer charge density with a cationic composition Zn2Al was preferentially chosen because the in situ polymerisation of interleaved monomers is known to be completely achieved.17,18 The nitrogen adsorption–desorption isotherm is displayed in Fig. 1(a) for this hybrid VBS/LDH material after in situ polymerisation and before any other thermal treatment. The absence of a noticeable microporosity is in agreement with the presence of highly packed polymer between the LDH sheets. After calcination at 600 °C but before demineralisation, some micropores are formed as observed by the increase of the nitrogen adsorbed volume in the low relative pressure domain (P/P0 < 0.1) (Fig. 1(b)). At this step, the sample is composed from aggregation of small particles of ZnS-like structure and Zn–Al–O based domains embedded into a carbon matrix.34 Therefore one should not expect neither a layered structure or a highly anisotropic character for the resulting carbon replica. | ||
| Fig. 1 N2 adsorption–desorption isotherms for the hybrid material after (a) in situ polymerization and (b) carbonisation. | ||
After acid-leaching of the various composites formed by pyrolysis under inert atmosphere, a steep rise in the adsorption curves is observed in the low relative pressure range (Fig. 2), showing that the acid treatment is liberating the micropores in the sample. By comparison with Fig. 1 for the carbons obtained after confinement, it underlines the fact that even if the water and gas release is initially acting as pore former, the elimination of the inorganic part is also of great importance.
 | ||
| Fig. 2 N2 adsorption–desorption isotherms for the different samples (1) C-VBS, (2) C-PSS, (3) C-ZVBS, (4) C-ZVBSpol and (5) C-ZPSS. | ||
The specific surface area obtained for the various samples are given in Table 1. From the Langmuir equation, the values are slightly overestimated probably due to the bimodal, micro- and mesoporosity present in the materials. Indeed, for the carbons obtained after constraint, the adsorption data above P/P0 of 0.1 (above the monolayer formation) deviate from flat to curved feature underlining that there is a significant sorption uptake on the external surface. After carbonization of the polymer PSS constrained or not (C-PSS and C-ZPSS, respectively), the values of specific area are in agreement with previous published results.13 However, in our case, much greater values are obtained when constrained monomers are used instead of polymers (i.e. C-ZVBS instead of C-ZPSS), and this is even more pronounced after the in situ polymerisation process (i.e. C-ZVBSpol gives greater values than C-ZVBS). The effect of air pre-treatment which is found to strongly orientate the textural properties may be questionable. However, based on 13C CP-MAS NMR results, we observed that the polymerisation of interleaved styrene sulfonate is an air assisted reaction since the process is not complete under nitrogen.18 Hence, since the LDH host structure is similar for all the samples, the differences of textural properties are believed to be due to differences in the polymer chain formation depending on the atmosphere. Indeed, in the absence of polymerisation and using 1,5-naphthalene disulfonate dianions (NDS), Puteyra et al. observed higher micropore surface area for the calcined organic molecular ions than for the carbon obtained after incorporation of the same molecule into the LDH gap structure.16
| Sample | V mono/cm3 g−1 (STP) | S BET/m2 g−1 | S L/m2 g−1 | S DK/m2 g−1 |
|---|---|---|---|---|
| a Hybrid material VBS/Zn2Al after in situ polymerisation b Hybrid material after carbonisation but before demineralisation | ||||
| Hybrida | 0.4 | 2 | — | — |
| After N2(T)b | 20.9 | 91 | 114 | 110 |
| C-VBS | 50.9 | 222 | 355 | 292 |
| C-PSS | 85.5 | 372 | 440 | 469 |
| C-ZVBS | 384 | 1671 | 1992 | 1719 |
| C-VBSpol | 513.0 | 2233 | 3560 | 2204 |
| C-ZPSS | 254.0 | 1105 | 1317 | 1177 |
The “discrete” distribution of micropores was carefully analyzed using three methods, t-plot, αs and Dubinin–Astakhov (D. A.) (Fig. 3). A greater contribution of micropores is obtained for carbons after sequestration into the LDH gap. The average size of the micropores, noted as Øμpore in Table 2 and visualized by the intercept between the dashed lines, is varying and the distribution in size as well. In the case of carbons prepared by sequestration, the distribution is found to be extended to values in the limit of attribution between micropores and mesopores. It is in agreement with previous observations where the carbon samples prepared by template carbonisation were found to present a heterogeneous microporous texture.13
 | ||
| Fig. 3 Analysis of the microporosity of the different samples by (a) t-plot, (b) αs and (c) Dubinin–Astakhov methods. Same notation as in Fig. 2. | ||
| Sample | t-plot | α s | Dubinin–Astakhov | V total | V μ/total d | |||
|---|---|---|---|---|---|---|---|---|
| S T/m2 g−1 (Øμpore/Å) | S N/m2 g−1 | V μpores c/mL g−1 (αs) | S ext/m2 g−1 | V μpores c/mL g−1 (P/P0) | βE0/kJ mol−1 | /mL g−1 (STP) | (%) | |
| a Hybrid material VBS/Zn2Al after in situ polymerisation b Hybrid material after carbonisation but before demineralisation c A detailed analysis is provided for the calculation of the microporous volume according to the choice of the pressure range. d V μ/total define the proportion of microporous volume, the lower number being considered as the proportion of smaller micropores. | ||||||||
| Hybrida | — | — | — | — | — | — | 4.14 | — |
| After N2(T)b | 123 (6.0–12.2) | — | 0.038 (0.6–2.0) | 4 | 0.039 (8.3 × 10−3–0.2) | 5.13 | 30.31 (0.047) | 81 |
| C-VBS | 303 (5.6–10.5) | — | 0.096 (0.7–2) | 20 | 0.103 (6 × 10−3–0.13) | 4.48 | 113.7 (0.176) | 59 |
| C-PSS | 331 (8.5–12) | 54 | 0.13 (0.07–0.4) | 125 | 0.168 (8.3 × 10−3–0.2) | 5.78 | 233.9 (0.360) | 47 |
| 0.16 (0.6–1.8) | 32 | |||||||
| C-ZVBS | 1478 (9.4–19.3) | 594 | 0.66 (0.07–0.5) | 663 | 0.65 (1 × 10−4–1.5·10−2) | 5.04 | 693.4 (1.07) | 62–68 |
| 0.73 (0.8–1.7) | 206 | |||||||
| C-ZVBSpol | 2166 (9.5–17.5) | 1322 | 0.511 (0.05–0.3) | 1435 | 0.78 (4.3 × 10−2–2 × 10−4) | 4.98 | 1103.3 (1.7) | 30–67 |
| 1.027 (0.7–2) | 145 | 1.14 (0.07–0.5) | 2.54 | |||||
| C-ZPSS | 1006 (8.2–17.6) | 374 | 0.317 (0.07–0.2) | 526 | 0.42 (1.4 × 10−4–4 × 10−2) | 5.79 | 511.9 (0.792) | 40–60 |
| 0.48 (0.7–1.8) | 197 | |||||||
A specific surface area attributed to narrow micropores (denoted SN) was calculated by the t-plot method (Table 2). For the αs method, downward curvatures on the curve showing the adsorbed volume Va(t) against αs was observed in the domain 0.2 < αs < 0.5, and for D–A, where the adsorption energy is governed by the pore size distribution, a deviation to linearity was depicted in the low relative pressure 10−4 < P/P0 < 10−1. The methods are consistent between each other, as larger micropore size distribution observed on the t-plot corresponds to the presence of either filling swings or changes of slope on the αs and D–A curves, respectively. The micropore volume between the different analyses is similar (Table 2). For example, for the sample C-ZPSS, micropores of small size contribute to 0.32 mL g−1, that corresponds 40% to the total adsorbed volume, whereas the total number of micropores represents 60%. The deviation observed on the αs curves above αs > 0.4 may be the result of pores larger than 1.5 nm, and, above 0.8, the upward deviation from linearity is then characteristic of mesopores.
For all the samples, the desorption curve is superimposable to the adsorption curve below a certain relative pressure. This value is dependent on the sizes of the mesopores.
For the samples C-ZVBS and C-ZPSS, the desorption curve is closing at P/P0 = 0.42; this corresponds to the capillary condensation of nitrogen into small mesopores, whereas for C-ZVBSpol, the hysteresis is closed at much higher relative pressure characteristic in this case of the presence of large sized mesopores. From the BJH method, a relatively narrow distribution is observed in this case with a mesopore size centered at 30 nm (Fig. 4). This value is comparable to the characteristics of other carbons obtained by colloid-imprinted35 or templating36 techniques and using mesoporous silica as the sacrifical host structure.
 | ||
| Fig. 4 Pore size distribution of the sample C-ZVBSpol. | ||
3.2. Raman study
Raman spectra recorded for the different samples are displayed in Fig. 5 together with spectra corresponding to Super S® and a commercial graphite that are also presented for the sake of comparison. | ||
| Fig. 5 Raman spectra of different samples (a) C-VBS, (b) C-PSS, (c) C-ZVBSpol, (d) C-ZVBS and (e) C-ZPSS. Graphite and Super S® Raman spectra are displayed for comparison. | ||
While the spectra of the as-made carbon materials exhibit barely no differences, two main vibrational bands are always observed in the 800–2000 cm−1 region as expected for carbon materials.37,38 The broad band labelled G and centered around 1600 cm−1 corresponds to the E2g symmetric vibrational mode of graphite-type sp2 carbons. The broad band labeled D around 1350 cm−1 is an indicator of structural disorder level characterized by bond angle distribution and linking with sp3 carbons. The co-existence of these two bands in our materials demonstrates their amorphous nature. Due to its preparation procedure, acetylene black can be considered as a reference for highly disordered carbons whereas graphite is a well ordered carbon structure. The peak features of the as-made carbons lie in between these two extremes (Fig. 5).
The intensity of the D and G Raman bands is often used to provide the microcrystalline planar size La of carbon materials, according to the relationship ID/IG = 4.4/La (nm).22,39,40 In our case, the values obtained for all the samples are in the lower limit of validity (La < 2.5 nm), thus underlining the presence of high degree of disorder. This is in agreement with both TEM analysis, where small and poorly ordered nanostructures were observed (not shown), and the XRD technique, where diagrams characteristic of amorphous materials were recorded. Moreover, the chemical analyses on the different carbon replicas show that the dissolution of the inorganic part was mostly complete (less than 0.5 wt% in total).
3.3. Capacitance properties
Since the carbon materials present a well balanced micro/mesoporosity, they were tested as electrodes for supercapacitors in 1 mol L−1 H2SO4. The ions from the electrolyte are adsorbed in the micropores, whereas mesopores are necessary to favor their quick diffusion to the active surface of micropores.41Fig. 6 presents the cyclic voltammograms (CV) for all the carbons in 1 mol L−1 H2SO4 measured at scan rates of 2 and 10 mV s−1. It can be observed that the CV curves prepared in the absence of constraint (samples C-VBS and C-PSS in Fig. 6 (a) and (b), respectively), present the characteristic box-like shape of an ideal capacitor even at moderate scan rates of 10 mV s−1. For the carbons prepared after LDH constraint (Fig. 6 (c), (d) and (e)), the CV curves present a less rectangular response, especially in the case of the sample C-ZVBSpol. This effect is more pronounced at higher scan rates of 10 mV s−1, that indicates some limitations in the charge transfer. One may surmise that due to the high proportion of mesopores, the carbon replicas are more resistive than the carbons prepared in the absence of structural constraint. Table 3 displays the specific capacitance, expressed by the mass of carbon of one electrode, calculated from the galvanostatic charge–discharge cycling experiments using a current density of 10 mA g−1. While a noticeable difference in the specific surface area is observed whether carbons are prepared with or without LDH constraint, the specific capacitance seems to be less correlated to the preparation. Consequently, the values of capacitance per surface unit, expressed in µF cm−2, indicate the best performance for the low surface area carbons prepared without constraint. One has to note that the capacitance for our samples is slightly underestimated as the mass of the composite electrode is considered in the calculation and that Super S® displays a very small gravimetric capacitance (Table 3).
 | ||
| Fig. 6 Cyclic voltammograms curves for the samples (a) C-VBS, (b) C-PSS, (c) C-ZVBSpol, (d) C-ZVBS and (e) C-ZPSS. Scan rate of 2 and 10 mV s−1. | ||
Usually, with porous carbons, it is considered that the charge is stored mainly in the double layer at the electrode/electrolyte interface. Therefore, many attempts try to relate the capacitance to the specific surface area, demonstrating generally a poor correlation.42–45 In some cases, the capacitance is found to be influenced by the presence of heteroatoms as observed in melamine-based carbons.46 After considering the data presented in Tables 2 and 3, we also remark that there is no clear relationship between capacitance and the nanotextural characteristics of the materials, as the surface area or the micropore volume. Therefore, other parameters such as the electrical conductivity of the materials or a possible contribution of pseudocapacitance due to the presence of surface groups involved in redox reactions, should be considered.
These results are confirmed by complex impedance spectroscopy experiments. Fig. 7 shows that at low frequency the imaginary part of the impedance curves approaches a vertical line in all cases, indicating a capacitive-type behavior. The carbons prepared without constraint present a phase angle close to π/2, indicating a capacitive behavior better than the carbons prepared by 2D constraint, in agreement with the CV data presented in Fig. 6. The intercept of the dashed lines to the Z′-axis corresponds to the Equivalent Serial Resistance (ESR) of the two-electrode capacitor (enlarged part of Fig. 7), which is due to the ionic resistivity of the electrolyte present in the porosity of the active material. From the ESR values collected in Table 3, one can remark that the carbons prepared without sequestration are less resistive than the carbons prepared from the same precursor by LHD constraint. The difference is remarkably higher when comparing carbons prepared from VBS than from those prepared from PSS. To explain the loss in the electrical conductivity, one may invoke the more open texture obtained from sequestration.
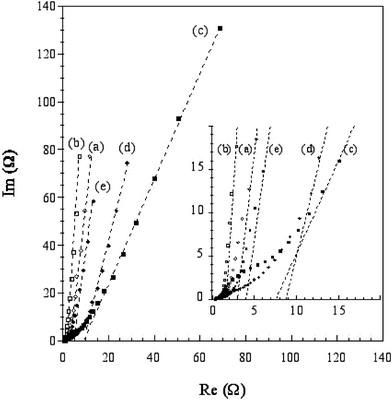 | ||
| Fig. 7 Complex plane impedance curves for the samples. Same notation as in Fig. 6. | ||
4. Conclusion
From the nitrogen adsorption–desorption analysis, we demonstrate that the initial hybrid formation has a strong effect on the textural properties, i.e. incorporation of monomer and its subsequent in situ polymerisation gives rise to higher micropore and mesopore contributions. Although, from the Raman study, it appears that the carbon materials present a similar disordered microstructure, made of very small microcrystalline planes. Characterized as EDLC electrode materials, the carbons exhibit moderate gravimetric capacitances. Carbons obtained after sequestration present a more pronounced pseudo capacitive character than those formed without using sequestration. Finally, it appears that for this family of compounds there is no apparent correlation between the textural properties, the carbon nano- and micro-structures and the behavior as electrochemical supercapacitors.In spite of their low performance as EDLC electrode materials, these carbons should be investigated for hydrogen energy storage.
Acknowledgements
The authors would like to thank Dr. El Mostafa Moujahid (LMI) for the preparation of some carbon replica and ENSCCF (Aubière) for the use of the nitrogen gas adsorption equipment.References
- M. T. Gilbert, J. H. Knox and B. Kavr, Chromatographia, 1982, 16, 138 CAS.
- T. Kyotani, N. Sonobe and A. Tomita, Nature, 1988, 28, 331 CrossRef.
- A. Bakandritsos, Th. Steriotis and D. Petridis, Chem. Mater., 2004, 16, 1551 CrossRef CAS.
- T. J. Bandosz, K. Putyera, J. Jagiello, J. A. Schwarz, J.-N. Rouzaud, F. Béguin and I. B. Maimoun, J. Chem. Soc., Faraday Trans., 1995, 91, 493 RSC.
- F. Béguin, J.-N. Rouzaud, I. B. Maimoun and A. Seron, J. Phys. Chem. Solids, 1996, 57, 1019 CrossRef CAS.
- Y. Xia and R. Mokaya, Chem. Mater., 2005, 17, 1553 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743 CrossRef CAS.
- D. Kawashima, T. Aihara, Y. Kobayashi, T. Kyotani and A. Tomita, Chem. Mater., 2000, 12, 3397 CrossRef CAS.
- H. Yang, Q. Shi, X. Liu, S. Xie, D. Jiang, F. Zhang, C. Yu, B. Tu and D. Zhao, Chem. Commun., 2002, 2842 RSC.
- A. B. Fuertes, J. Mater. Chem., 2003, 13, 3085 RSC.
- A. Macario, A. Katovic, G. Giordano, F. Iucolano and D. Caputo, Microporous Mesoporous Mater., 2005, 81, 139 Search PubMed.
- Y. Ma, Z. Kyotani and T. A. Tomita, Carbon, 2002, 40, 2367 CrossRef CAS.
- K. Putyera, T. J. Bandosz, J. Jagiello and J. A. Schwarz, Carbon, 1996, 34, 1559 CrossRef CAS.
- T. Bandosz, J. Jagiello, K. Putyera and J. A. Schwarz, Chem. Mater., 1996, 8, 2023 CrossRef CAS.
- K. Putyera, T. J. Bandosz, J. Jagiello and J. Schwarz, Applied Clay Sci., 1995, 10, 177 Search PubMed.
- K. Putyera, T. J. Bandosz, J. Jagiello and J. A. Schwarz, Clays Clay Miner., 1994, 42, 1 CrossRef CAS.
- L. Vieille, C. Taviot-Guého, J.-P. Besse and F. Leroux, Chem. Mater., 2003, 15, 4369 CrossRef CAS.
- M. El. Moujahid, J.-P. Besse and F. Leroux, J. Mater. Chem., 2002, 12, 3324 RSC.
- F. Leroux and J.-P. Besse, Chem. Mater., 2001, 13, 3507 CrossRef CAS.
- F. Leroux and C. Taviot-Guého, J. Mater. Chem., 2005, 15, 3628 RSC.
- A. C. Ferrari and J. Robertson, Phys. Rev. B, 2000, 61, 14095 CrossRef CAS.
- P. Lespade, A. Marchand, M. Couzi and F. Cruege, Carbon, 1984, 22, 375 CAS.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937 CrossRef CAS.
- K. Jurewicz, C. Vix-Guterl, E. Frackowiak, S. Saadallah, S. Reda, J. Parmentier, J. Patarin and F. Béguin, J. Phys. Chem. Solids, 2004, 65, 287 CrossRef CAS.
- C. Vix-Guterl, S. Saadallah, K. Jurewicz, E. Frackowiak, M. Reda, J. Parmentier, J. Patarin and F. Béguin, Mat. Sci. Engineering, 2004, 108, 148 Search PubMed.
- C. Vix-Guterl, E. Frackowiak, K. Jurewicz, M. Friebe, J. Parmentier and F. Béguin, Carbon, 2005, 43, 1293 CrossRef CAS.
- J. H. de Boer, B. G. Linsen, T. van der Plas and G. J. Zondervan, J. Catalysis, 1965, 4, 649 Search PubMed.
- J. H. de Boer, B. C. Lippens, B. G. Linsen, J. C. P. Broekhoff, A. van den Heuvel and Th. J. Osinga, J. Colloid Interface Sci., 1966, 21, 405 CAS.
- M. Kruk, M. Jaroniec and A. Sayari, Langmuir, 1997, 13, 6267 CrossRef CAS.
- M. Kruk, M. Jaroniec and K. P. Gadkaree, J. Colloid Interface Sci., 1997, 192, 250 CrossRef CAS.
- N. D. Hutson, D. J. Gualdoni and R. T. Yang, Chem. Mater., 1998, 10, 3707 CrossRef CAS.
- Z.-M. Wang, Z. Liu, N. Yamashita, H. Kanoh and K. Ooi, Langmuir, 2002, 18, 1957 CrossRef CAS.
- M. Kruk, M. Jaroniec, T.-W. Kim and R. Ryoo, Chem. Mater., 2003, 15, 2815 CrossRef CAS.
- M. El Moujahid, J.-P. Besse and F. Leroux, J. Mater. Chem., 2003, 13, 258 RSC.
- Z. Li and M. Jaroniec, Chem. Mater., 2003, 15, 1327 CrossRef CAS.
- C.-M. Yang, C. Weidenthaler, B. Spliethoff, M. Mayanna and F. Schüth, Chem. Mater., 2005, 17, 355 CrossRef CAS.
- R. Kostecki, T. Tran, X. Song, K. Kinoshita and K. McLarnon, J. Electrochem. Soc., 1997, 144, 3111 CAS.
- M. J. Matthews, M. S. Dresselhaus, M. Endo, Y. Sasabe, T. Takahashi and K. Takeuchi, J. Mater. Res., 1996, 11, 3099 CrossRef CAS.
- N. Shimodaira and A. Masui, J. Appl. Phys., 2002, 92, 902 CrossRef CAS.
- T. V. Reshetenko, L. B. Avdeeva, Z. R. Ismagilov, V. V. Pushkarev, S. V. Cherepanova, A. L. Chuvilin and V. A. Likholobov, Carbon, 2003, 41, 1605 CrossRef CAS.
- L. Duclaux, E. Frackowiak, T. Gibinski, R. Benoit and F. Béguin, Mol. Cryst. Liq. Cryst., 2000, 340, 449 CrossRef CAS.
- Y. J. Kim, Y. Horie, S. Ozaki, Y. Matsuzawa, H. Suezaki, C. Kim, N. Miyashita and M. Endo, Carbon, 2004, 42, 1491 CrossRef CAS.
- M. Endo, Y. J. Kim, T. Takeda, T. Maeda, T. Hayashi, K. Koshiba, H. Hara and M. S. Dresselhaus, J. Electochem. Soc., 2001, 148, A1135 Search PubMed.
- M. Endo, Y. J. Kim, K. Ishii, T. Inoue, T. Takeda, T. Maeda, T. Nomura, N. Miyashita and M. S. Dresselhaus, J. Electrochem. Soc., 2002, 149, A1473 CrossRef CAS.
- D. Qu and H. Shi, J. Power Sources, 1998, 74, 99 CrossRef CAS.
- D. Hulicova, J. Yamashita, Y. Soneda, H. Hatori and M. Kodama, Chem. Mater., 2005, 17, 1241 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM pictures of C-VBS, C-PSS, C-ZPSSpol and C-ZPSS; cyclic voltammograms of Super S, scan rate of 2 and 10 mV s−1; complex plane impedance curves for Super S; D and G contribution from the Raman spectrum analysis. See DOI: 10.1039/b600513f |
| This journal is © The Royal Society of Chemistry 2006 |