Alternate copolymers of head to head coupled dialkylbithiophenes and oligoaniline substituted thiophenes: preparation, electrochemical and spectroelectrochemical properties†
K.
Buga
a,
R.
Pokrop
a,
A.
Majkowska
a,
M.
Zagorska
*a,
J.
Planes
b,
F.
Genoud
b and
A.
Pron
b
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland.
bDRFMC, UMR 5819 SPrAM (CEA–CNRS–Université J. Fourier Grenoble I), Laboratoire Electronique Moléculaire, Organique et Hybride, CEA Grenoble, 17 rue des Martyrs, 38054 Grenoble Cedex 9, France.
First published on 10th April 2006
Abstract
New processable, electroactive, alternate copolymers consisting of dialkylbithiophene units and oligoanilinethiophene units have been prepared by post-polymerization functionalization of a specially prepared precursor polymer, namely poly[(4,4″-dioctyl-2,2′∶5′,2″-terthiophene-3′-yl)ethyl acetate], carried out via its hydrolysis and consecutive branching aniline dimer or tetramer through the amidation reaction. The precursor polymer is interesting by itself because it gives a very clear spectroelectrochemical response over a very narrow potential range. The proposed method enables the preparation of regiochemically better defined alkylthiophene-oligoanilinethiophene copolymers with higher content of oligoaniline side groups as compared to previously used methods. Cyclic voltammetry investigations combined with UV–vis–NIR, EPR and Raman spectroelectrochemistry show that both the oligoaniline side groups and poly(thienylene) main chain are electrochemically active. Significant differences for the side group electrochemistry are observed in acidified and nonacidified electrolytes making the prepared new copolymer a good candidate for electrochromic applications in diversified electrolytes.
Introduction
Post-polymerization functionalization of conjugated polymers constitutes a convenient tool for the preparation of new electroactive organic systems with tunable electronic, electrochemical and spectroscopic properties.1,2 A typical approach used in the preparation of such polymers consists of the synthesis of specially designed polymerizable macromonomers containing processing improving substituents and one or more functional groups which serve as grafting sites in the post-functionalization procedure.3 The polythiophene family of conjugated polymers has been especially interesting in this respect. First, thiophene based macromonomers can be relatively easily polymerized using either chemical or electrochemical oxidative polymerization1 or via different types of polycondensation methods including Kumada, Stille, Suzuki or Yamamoto coupling, the details on these reactions can be found in an excellent review of Miyaura.4 Second, for polythiophene and its derivatives the introduction of solubility inducing side groups does not influence, in the negative sense, their electroactivity. Finally, in the majority of cases, the post-functionalization enables a controllable tuning of several important properties of these systems while retaining their solution processibility.5 In recent years, the post-polymerization functionalization has been used for branching to the polythiophene main chain chromophores exhibiting second order non-linear optical properties,6,7 metal complexing sites8 or electrochemically active groups,2,9–13 to name a few.In this perspective, the post-polymerization functionalization seems a very suitable method for the preparation of “hybrid” polymers containing a polythiophene main chain and oligoaniline side chains. The interest in this type of polymers arises mainly from their interesting electrochemistry and spectroelectrochemisty. Since the branched oligoaniline substituents mimic the behaviour of polyaniline, they are electrochemically active both in non-aqueous and aqueous electrolytes, in the latter case their electrochemical response being pH dependent. The electrochemical behaviour of the main chain resembles, in turn, that of polythiophenes. Thus by an appropriate combination of redox and/or acid base reactions it is possible to dope these polymers either selectively in their side chains or globally in the side chains and in the main chain.
In one of our previous works we have prepared poly(alkylthiophene) containing side oligoaniline groups by copolymerization of 3-octylthiophene and 3-oligoanilinethiophene.14 The principal disadvantage of this method is associated with the fact that the resulting system is a random copolymer with irregular distribution of both alkyl and oligoaniline substituents along the chain. Moreover, because of a large difference in the reactivity between both comonomers with respect to Cα–Cα′ coupling only small numbers of oligoaniline groups can be introduced to the resulting copolymer. In the second method, described in the papers of Buga et al.15,16 we succeeded in the preparation of copolymers with much higher oligoanilinethiophene to alkylthiophene ratios but the systems obtained were still random copolymers with a chain microstructure that is impossible to control.
It seems therefore interesting to prepare polymers with a higher content of oligoaniline side chains and a more regular chain structure. This is a problem of crucial importance since spectroscopic and electrochemical properties of polythiophenes depend not only on the degree of the regioregularity but also on its type. For example polymerization of two macromonomers which are positional isomers may lead to distinctly different properties of the resulting polymers because of the different type of the chain regioregularity imposed in each case.17
Substituted terthiophenes of different symmetry constitute convenient substrates for the preparation of regioregular polymers which can then be post-functionalized.18,19 Using this approach we present here the synthesis of ester group substituted poly(4,4″-dialkyl-2,2′∶5′,2″-terthiophene) which then serves as a precursor polymer for the grafting of oligoanilines. The post-functionalization of the precursor polymer is achieved through a sequence of reactions consisting of the hydrolysis of the ester groups followed by aniline dimer or tetramer attachment via amidation reaction. Both the precursor and the post-functionalized polymers exhibit very interesting electrochromic, electrochemical and spectroelectrochemical (UV–vis, Raman and EPR) properties.
Experimental
Preparation of the precursor polymer (P1)
Details concerning the reagents used, their purification and the synthesis of the macromonomer (3) (Scheme 1) can be found in the electronic supplementary information (ESI).† | ||
| Scheme 1 | ||
The precursor polymer P1 was obtained via oxidative polymerization of the trimer (4,4″-dioctyl-2,2′;5′,2″-terthiophene-3′-yl)ethyl acetate (3), using anhydrous FeCl3 as the oxidizing–polymerizing agent in a procedure similar to that described in20 (see Scheme 2). In a typical polymerization 0.93 g (5.7 mmol) of FeCl3 in 30 mL of dry nitromethane were slowly added under argon flow over ca. 15 min to a solution of 0.80 g (1.4 mmol) of (3) in 20 mL of dry CCl4. After the addition of the oxidant the reaction mixture turned brown. The reaction was carried out for 2 h and then was terminated by precipitation of the polymer formed with methanol. The resulting rubbery product was repeatedly washed with methanol until the filtrate was colorless. As formed, crude polymer always contains residual dopants of a frequently unknown chemical nature and requires careful dedoping. The dedoping was carried out in an ammonia solution in methanol (1 ∶ 5) for 2 h and was followed by soaking the polymer in pure methanol for 24 h. Finally the dedoped polymer was dried to a constant mass yielding 0.78 g of the product (yield 97.5%).
 | ||
| Scheme 2 | ||
1H NMR (CDCl3, ppm): 7.11–7.05 (m, 3H); 4.18–4.17 (m, 2H); 3.79–3.74 (m, 2H); 2.57–2.51 (m, 4H); 1.62–1.55 (m, 4H); 1.30–1.24 (m, 23H); 0.85–0.84 (m, 6H); FTIR (cast on KBr pellet, cm−1): 3057 (w), 2956 (vs), 2926 (vs), 2855 (vs), 2724 (w), 1739 (s), 1653 (w) 1560 (w), 1521 (w), 1464 (m), 1378 (m), 1367 (m), 1319 (m), 1256 (m), 1182 (m), 1159 (m), 1127 (w), 1097 (w), 1034 (m), 830 (m), 723 (w); UV–VIS (CHCl3 solution, nm) : 406, 254. Elemental analysis: calcd for C32H44S3O2: C, 69.06%; H, 7.91%; S, 17.26%. Found: C, 68.26%; H, 7.77%; S, 17.33%.
Post-polymerization functionalization of the precursor polymer
The post-polymerization functionalization of the precursor polymer (P1) was carried out in two steps. First, the precursor P1 was hydrolyzed to give the corresponding polyacid (P2), then P3 and P4 were obtained via amidation reaction between the carboxylic groups of P2 and the primary amine groups of dianiline and tetraaniline, respectively.Hydrolysis of P1 to give P2. The applied hydrolysis procedure was a modification of the method described in.21 The applied procedure can briefly be described as follows. 0.1 g of the precursor polymer (P1) was dissolved in 5 mL of CHCl3, then 10 mL of 2 M NaOH solution in EtOH were added dropwise. The mixture was then heated to the boiling temperature and kept at this temperature for 24 h under reflux. The product in a form of a yellow suspension was separated by filtration on a Büchner funnel, stirred in 2 M HCl–methanol solution (5 ∶ 1) for few hours and finally vacuum-dried. 1H NMR (CDCl3, ppm): 7.11–6.93 (m, 3H); 3.81–3.69 (m, 2H); 2.58–2.48 (m, 4H); 1.56–1.22 (m, 24H); 0.84–0.83 (m, 6H); FTIR (KBr, cm−1): 3055 (w), 2953 (s), 2925 (vs), 2853 (s), 2676 (w), 1710 (s), 1520 (w), 1463 (m), 1412 (w), 1377 (m), 1300 (m), 1214 (m), 1032 (w), 934 (w), 828 (m), 722 (w).
Preparation of P3 and P4 from P2 and oligoanilines. The reaction of aniline dimer or tetramer grafting was carried out under dry argon flow using a procedure frequently applied to the esterification or amidation reactions.22
In the preparation of P3, 80 mg of the hydrolyzed precursor polymer (P2) were dissolved in 10 mL of dry THF. Solution of 28 mg (0.15 mmol) of aniline dimer in 10 mL of dry THF, 31 mg (0.15 mmol) of DCC and 2 mg (0.02 mmol) of DMAP were consecutively added. The reaction was stopped after 24 h and the reaction mixture was filtered to remove reaction side products. In the next step THF was pumped off on a rotary evaporator and the remaining polymer was repeatedly washed with methanol until the filtrate was colorless. Finally the polymer powder obtained was dried in a vacuum line to a constant mass. 1H NMR (CDCl3, ppm): 7.35–6.74 (m, 12H); 3.79–3.66 (m, 2H); 2.60–2.41 (m, 4H); 1.77–1.48 (m, 4H); 1.40–1.00 (m, 20H); 0.87–0.70 (m, 6H); FTIR (KBr, cm−1): 3313 (w), 3055 (w), 2927 (vs), 2855 (vs), 1690 (s), 1658 (s), 1598 (s), 1516 (vs), 1497 (s), 1465 (m), 1317 (m), 1257 (m), 831 (m), 746 (m), 734 (m). UV–VIS (CHCl3 solution, nm): 409, 300 nm. Elemental analysis: calcd for C42H50N2S3O·2H2O: C, 69.04%; H, 7.39%; N, 3.83%; S, 13.15 Found: C, 69.60%; H, 7.40%; N, 3.82%; S, 13.15%.
In the case of the synthesis of P4, a solution of 69 mg (0.19 mmol) of aniline tetramer in 10 ml of dry THF, 39 mg (0.19 mmol) of DCC and 2 mg (0.02 mmol) of DMAP were consecutively added to 0.1 g of the hydrolyzed precursor polymer (P2) dissolved in 10 mL of dry THF. The reaction was stopped after 24 h. The mixture was filtered, in order to remove reaction side products. In the next step THF was removed on a rotary evaporator and the remaining polymer was repeatedly washed with methanol until the filtrate was colorless. Finally the obtained polymer P4 powder was dried in a vacuum line till constant mass. 1H NMR (CDCl3, ppm): 7.35–6.74 (m, 20H); 3.79–3.66 (m, 2H); 2.56–2.41 (m, 4H); 1.67–1.48 (m, 4H); 1.28–1.20 (m, 20H); 0.87–0.81 (m, 6H); FTIR (KBr, cm−1): 3365 (m), 3055 (w), 2923 (vs), 2852 (s), 2676 (w), 1740 (m), 1707 (m), 1660 (m), 1591 (s), 1510 (vs), 1496 (vs), 1465 (m), 1453 (m), 1377 (m), 1314 (s), 1259 (m), 1211 (m), 1168 (m), 1129 (m), 1076 (m), 1027 (m), 831 (m), 746 (w), 722 (w), 695 (w); UV–vis (CHCl3 solution, nm): 558, 403, 311 nm. Elemental analysis: calcd for C54H58N4S3O·3H2O: C, 69.83%; H, 6.90%; N, 6.03%. Found: C, 70.07%; H, 7.00%; N, 6.02%.
Characterization techniques
The trimer used for the polymerization (3) was characterized by 1H NMR, 13C NMR, FTIR, mass spectrometry and elemental analysis, the precursor polymer (P1), its hydrolyzed form (P2) and the final post-functionalized polymers containing oligoaniline side groups (P3 and P4) were identified by 1H NMR, FTIR and subjected to combustion analysis.NMR spectra were recorded on a Varian Mercury 400 MHz spectrometer with chloroform-d (CDCl3) solvent.
FTIR spectra were recorded on a FTIR Bio-RAD FTS 165 spectrometer (wavenumber range: 400–4000 cm−1, resolution: 4 cm−1) either on free standing films cast from chloroform or using the KBr pellet technique. UV–vis–NIR spectra of thin solid films or solutions in chloroform were recorded on a Lambda 2 Perkin Elmer spectrometer.
Molecular weight determinations of the synthesized polymers were measured using size exclusion chromatography (SEC) on a Shimadzu LC–10 AD chromatograph equipped with a Nucleogel M-10 column, using refractometric detection by RID-6A refractometer. THF was used as the eluent. The column temperature and the flow rate were fixed to 35 °C and 1 mL min−1, respectively. The column was calibrated using polystyrene standards provided by Polymer Standards Service.
For cyclic voltammetry investigations a thin layer of the polymer was deposited on a platinum disc electrode by casting from chloroform solution. A typical surface concentration of the electroactive sites, estimated from coulometry (assuming 0.33 doping level), was ca. 1–2 × 10−8 mol cm−2. The experiments were carried out in a one compartment electrochemical cell using a Pt counter-electrode, and an Ag/0.1 M AgNO3 in acetonitrile reference electrode. Two types of electrolyte were used: 0.1M tetrabutylammonium tetrafluoroborate (TBATFB) or a mixture of 0.1 M TBATFB and 0.1 M diphenyl phosphate (DPP) in acetonitrile, the latter served as a source of protons facilitating the redox processes associated with the grafted aniline tetramer.14 To eliminate the so called “memory effect” few scans were performed before the measurements.
The same electrolytes as well as the reference and counter electrodes were used in the case of UV–vis–NIR, Raman and EPR spectroelectrochemical studies. In the case of UV–vis–NIR spectroelectrochemical studies a thin polymer film was deposited on an ITO transparent working electrode. Platinum electrodes in a form of a plate and a wire were used in Raman and EPR studies, respectively. The UV–Vis–NIR spectra were measured on a Lambda 2 Perkin Elmer spectrometer whereas the Raman spectra were obtained using a FT Raman Brücker RFS 100 spectrometer with the near-IR excitation line (1064 nm). The EPR experiments were performed with an ER 200 Brücker X band spectrometer. The EPR susceptibility was obtained by double integration of the line which were normalized with respect of a commercial calibrated sample (strong pitch Brücker) placed in the same dual cavity.
Results and discussion
Synthesis
Several synthetic routes have been exploited in the past two decades for the preparation of functionalized polythiophenes. One of the most popular methods involves the preparation of specially designed macromonomers, which—depending on their symmetry—may lead to polymers of different regioregularity. The trimer (3), which upon oxidative coupling gives the precursor polymer (P1), can be obtained in good yields from 3-octylthiophene and ethyl-3-thiopheneacetate using the reaction sequence depicted in Scheme 1. The applied procedure consists of the halogenation of ethyl-3-thiopheneacetate with N-bromosuccinimide to give its dibromoderivative (2) and the transformation of 3-octylthiophene into its boronate derivative (1).23,24 Suzuki type coupling25–27 of 1 and 2, using palladium tetrakis triphenylphosphine (Pd[P(Ph)3]4) as the catalyst, gives the desired macromonomer (3). Chemical oxidative polymerization of 3 with FeCl3 as the polymerizing–oxidizing agent results in the formation of the precursor polymer (P1), in which octyl groups play the role of solubility inducing substituents whereas the ester group in the central ring, after its hydrolysis, serves as the site of oligoaniline grafting (see Scheme 2). The number average molecular weight of P1, Mn, determined by SEC using polystyrene standards, is 5.9 kDa with the polydisperisty index PI = 2.9. This corresponds to the polymerization degree, normalized for one 2,5-thienylene ring, DPn = 28. A rather high value of the PI is typical of thiophene derivatives polymerization using FeCl3.28 It should be noted here that both Mn and DPn values may be overestimated. Liu et al.29 by comparison of the Mn value determined from MALDI-TOF measurements with the corresponding value obtained from SEC using polystyrene standards, came to the conclusion that the SEC results are overestimated by ca. 30% for number average molecular weights in the range 5000–6000.Both elemental analysis (see Experimental) and 1H NMR results (see (ESI)) are consistent with the chemical constitution of P1 presented in Scheme 2. In particular, the ratio of the integrated signals at 0.88 ppm and at 4.18 ppm is 2.92 which is very close to the theoretical value of 3. The former corresponds to six protons of the two methyl groups in both alkyl substituents whereas the latter two protons of the methylene group are adjacent to oxygen in the ester substituent.
The reactions of the hydrolysis of P1 followed by grafting of an aniline dimer or tetramer to give P3 and P4, respectively, are quantitative as judged from the total disappearance of the 1H NMR signal at 4.18 ppm in the reaction products as well as from the elemental analysis. It should however be noted here that in both cases (P3 and P4) a good fit of the elemental analysis can be obtained only if one assumes that the hydrophilic components of the polymers are solvated with water molecules. This is not unexpected since both polyaniline and its oligomers are known to adsorb significant quantities of water which is difficult to totally remove even after extended pumping.14 In Fig. 1 the chloroform solution UV–vis spectrum of P1 is compared with those registered in the same solvent for P3 and P4. In the case of P1 the dominant peak at ca. 400 nm corresponds to the π–π* transition in the conjugated polymer backbone. This band is hypsochromically shifted with respect to the corresponding band in other substituted poly(thiophene)s. The observed shift reflects a significant chain torsion caused by the head to head (HH) coupling of the adjacent alkylthienylene rings.30 Branching of aniline dimer or tetramer via hydrolysis of the ester group followed by the amidation reaction practically does not influence this transition. However it gives rise to a new band at ca. 300–310 nm, which can be attributed to the π–π* transition in the phenylene ring in P3 and P4, respectively.14,15 The spectrum of P3 contains only these two, above mentioned, peaks which unequivocally show that the branched dimer is in its most reduced state. To the contrary, in P4 an additional band can be distinguished at about 580 nm. This peak is indicative of the semi-oxidized state of the grafted tetramer and is usually ascribed to an excitonic type transition between the HOMO orbital of the benzoid ring and the LUMO orbital of the quinoid one.31
 | ||
| Fig. 1 Solution (chloroform solvent) spectra of: (a) the precursor polymer P1 (solid line); the post-functionalized polymer containing aniline dimer side groups P3 (dotted line); (b) the post-functionalized polymer containing aniline tetramer side groups P4 (solid line); P4 after acidification with diphenyl phosphate (dotted line). | ||
The oxidation state of the grafted oligomers is further corroborated by spectral changes caused by the acidification of the polymer solution with diphenyl phosphate (DPP) (see Fig. 1b). It is known that polyaniline or its oligomers in the semi-oxidized form undergo protonation of the imine nitrogens followed by charge redistribution leading to the structure of a polysemiquinone radical. In the case of P4 the addition of DPP results in an immediate disappearance of the bands at 310 nm and at about 580 nm with concomitant growth of a new band peaked at 820 nm. All these changes are characteristic of the aniline tetramer side group protonation.32 To the contrary, the spectrum of P3 does not change upon addition of DPP since reduced forms of oligoaniline do not undergo protonation in these conditions.
Cyclic voltammetry and UV–vis–NIR spectroelectrochemistry
Poly(alkylthiophene)s containing head to head (HH) coupled alkylthienylene units show a very characteristic voltammetric behaviour with a very sharp and narrow anodic peak associated with the p-type doping and its much broader cathodic counterpart ascribed to the reduction of the oxidized (p-doped) polymer to the neutral state.33 A very similar cyclic voltammogram is registered for P1 (see Fig. 2a). The exchange of ester groups, which are electrochemically inactive in this potential range, for aniline dimers strongly influences the shape of the cyclic voltammogram (Fig. 2b). | ||
| Fig. 2 Cyclic voltammograms of: (a) the precursor polymer P1 (electrolyte composition 0.1 M Bu4NBF4 in acetonitrile, scan rate 50 mV s−1); (b) the post-functionalized polymer containing aniline dimer side groups P3 (electrolyte composition 0.1 M Bu4NBF4 in acetonitrile, scan rate 50 mV s−1) (c) the post-functionalized polymer containing aniline tetramer side groups P4 (electrolyte composition 0.1 M Bu4NBF4 in acetonitrile, scan rate 50 mV s−1); (d) the post-functionalized polymer containing aniline tetramer side groups P4 recorded in acidified electrolyte (electrolyte composition 0.1 M Bu4NBF4 + 0.1 M diphenyl phosphate in acetonitrile, scan rate 50 mV s−1). | ||
In the case of P3 a significant broadening of the principal anodic peak can be observed with a simultaneous shift of its maximum towards higher potentials. Moreover, a new broad peak, preceding the principal peak, appears at lower potentials. We ascribe this new peak to the oxidation of the reduced, neutral form of the branched dimer. This can occur in two steps with the formation of a radical cation in the first step and, after further oxidation and deprotonation in the second step, an iminequinone unit (see Scheme 3). This assignment is based on close similarity of the observed voltammetric behaviour to that reported for “free” aniline dimer34 and for the side group dimer in a random copolymer of alkylthiophene and oligoaniline thiophene.15 The spectroelectrochemical response of P3 is also consistent this mechanism (vide infra).
 | ||
| Scheme 3 Oxidation of the oligoaniline side chain. | ||
The observed voltammetric behaviour implies sequential doping, which involves the oxidative charging of the side groups at the potentials of the first anodic peak followed by the oxidative doping of the main chain in the potential range of the second anodic peak. Very similar voltammetric features are observed for P4 in which, similarly as in the previous case, the broad peak at lower potentials can be ascribed to the doping of the side oligoaniline groups whereas the principal peak to the doping of the main chain (Fig. 2c). These conclusions are also corroborated by complementary spectroelectrochemical investigations (vide infra).
A linear relationship between the peak current and the scan rate is found for all polymers studied both for the oxidative doping and reductive dedoping peaks, consistent with thin layer behaviour (see P3 as an example in the ESI†). The shape and the position of the peaks of the redox couple corresponding to the doping–dedoping of the main chain (second redox couple) do not depend on the scan rate. This is characteristic of conjugated polymers layers which become conductive in the doping process. To the contrary, the position of the maximum of the first oxidation peak, corresponding to the charging of the side oligoaniline groups is scan rate dependent. Evidently, macroscopic conductivity of the layer is not achieved at these potentials, since the charge imposed on the side groups cannot easily propagate without the participation of the main chain. Thus, this observation additionally corroborates the sequential character of the doping of polymers containing oligoaniline side chains. Note, that the position of the reduction peak of the first redox couple does not depend on the potential scan rate. This is not unexpected since in the reductive dedoping some residual charges remain both on the main chain and the side chains assuring some conductivity of the layer. These residual charges, together with some relaxation phenomena give rise to so called “memory effect” observed in the majority of conducting polymers.
In view of a clear spectroscopic indication of the side, tetramer group protonation of P4 in acidified electrolytes it seems interesting to verify the influence of the protonation process on the voltammetric and spectroelectrochemical behaviour of this polymer. In principle, the presence of an efficient protonating agent should facilitate all redox processes in which the electron transfer is accompanied by proton abstraction or addition. For this reason we have carried out detailed electrochemical and spectroelectrochemical studies of P4 in a mixed electrolyte consisting of equimolar amounts of a non-protonating (TBATFB) and a protonating (DPP) component in acetonitrile (0.1 M with respect to each component). An efficient reduction of the tetramer side groups of P4, in the DPP acidified electrolyte, can be conveniently monitored by the UV–vis spectroscopy (see Fig. 3). When the electrode potential is lowered from its open circuit value of Eoc = +0.20 V to E = −0.20 V, essentially all spectroscopic features, characteristic of the semi-oxidized, protonated form of the grafted aniline tetramer, disappear. The resulting spectrum consists of two peaks at 300 nm and about 400 nm, characteristic of the neutral, totally reduced form of the side aniline tetramer and of the neutral, undoped form of the poly(thienylene) main chain, respectively. No such reduction is observed for P4 in the nonacidified electrolyte. In this case the open circuit potential value is Eoc = −0.30 V and the UV–vis spectrum, registered at this potential, shows the presence of three peaks. The first one about 400 nm is characteristic of the reduced neutral main chain whereas the remaining two, at 300 and 580 nm, can be ascribed to the semi-oxidized tetraaniline side chains in their base form. The cyclic voltammogram of P4 registered in the acidified electrolyte resembles to a first approximation that registered in the non-acidified one, i.e. it consists of a broad peak at lower potentials ascribed to the oxidation of the tetraaniline side group and a more narrow peak at higher potentials attributed to the oxidation of the poly(thienylene) main chain (see Fig. 2d).
 | ||
| Fig. 3 UV–vis–NIR spectra of a thin solid film of P4 deposited on an ITO electrode registered in the reduction mode by lowering the potential from Eoc = +0.20 V to E = −0.20 V recorded in acidified electrolyte (electrolyte composition 0.1 M Bu4NBF4 + 0.1 M diphenyl phosphate in acetonitrile). | ||
Voltammetric behaviour of P1, P3 and P4 is reflected in their UV–vis spectroelectrochemical response. In Figs. 4a–d the UV–vis spectra recorded for increasing electrode potentials are presented for all polymers studied. Consistent with its cyclic voltammogram, for P1 no doping induced spectroscopic changes are observed up to E = +0.40 V. Then over the potential range of 25 mV a drastic decrease of the intensity of the band characteristic of the neutral polymer occurs (the band ascribed to the π–π* transition in the poly(thienylene) conjugated backbone at 403 nm). Simultaneously, two new, doping induced bands grow at higher wavelengths. The peak at higher energies gives rise to a maximum at 770 nm. Due to the spectrometer limit at 1100 nm only the onset of the second band is registered since its maximum is located in the near infrared. Note that at E = +0.70 V the doping is essentially completed as judged from the residual intensity of the band corresponding to the neutral polymer. Thus the spectroelectrochemical behaviour of P1 resembles that reported for HH-TT coupled regioregular poly(alkylthiophene)s and is governed by the difficulty of the charge removal caused by a significant non-planarity of the polymer chain.33
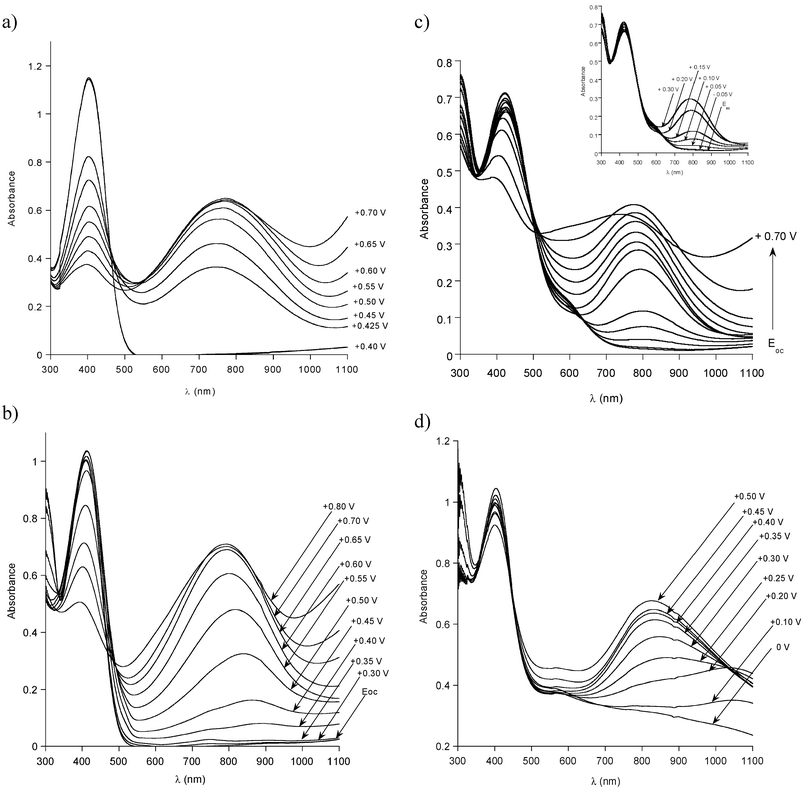 | ||
| Fig. 4 UV–vis–NIR spectra, registered for increasing electrode potentials (Evs. Ag/Ag+): (a) P1 in 0.1 M Bu4NBF4–acetonitrile solution; (b) P3 in 0.1 M Bu4NBF4–acetonitrile solution; (c) P4 in 0.1 M Bu4NBF4–acetonitrile solution; (d) P4 in (0.1 M Bu4NBF4 + 0.1 M diphenyl phosphate)–acetonitrile solution. | ||
P3 which is obtained from P1 through the grafting of aniline dimer starts to oxidize at lower potentials as compared to the case of P1. At the open circuit potential (Eoc = 0 V) two bands at 300 nm and 410 nm, can be distinguished, corresponding to the π–π* transitions in the aromatic ring of the dianiline side group and the poly(thienylene) main chain, respectively (Fig. 4b). No changes in the spectrum are observed until E = +0.30 V. Above this potential value a decrease in the intensity of the band at 300 nm is registered with essentially no changes in the intensity of the band at 410 nm. Taking into account a close similarity of these results to those obtained for random copolymers of alkylthiophene and thiophene substituted with dianiline,15 we are tempted to interpret it as the oxidation of the dianiline side chains to radical cations with the main chain remaining intact. The first features of the oxidation of the main chain appear at E = +0.45 V i.e. in the potential range where the two peaks present in the cyclic voltammogram overlap (see Fig. 2b). Our attribution of these changes to the main chain doping is based on the following observations: first, beginning from this potential value a steady decrease in the intensity of the band attributed to the π–π* transition in the thienylene ring can be observed with a correlated growth of the doping induced peak in the vicinity of 795 nm, which can be ascribed to the main chain doping. Second, in the same potential range only minimal changes in the intensity of the peak at 300 nm, corresponding to the oxidation of the side aniline dimer, are registered. At the high potential values the spectroelectrochemical behaviour of P3 resembles that of P1 corroborating the doping of the main chain. Of course spectral changes occur over a broader potential range in agreement with the cyclic voltammetry.
As it has already been stated (vide supra), in the non-acidified electrolyte, at Eoc = −0.30 V, aniline tetramer side groups in P4 are in the semi-oxidized, base state. The first changes in the spectrum of the polymer, induced by its oxidation, are observed at E = 0 V (Fig. 4c). In the potential zone covering the first, broad oxidation peak in the cyclic voltammogram, only the bands corresponding to the grafted tetramer are affected i.e. the peak at 300 nm and that at 580 nm (see the inset in Fig. 4c). Both become less intensive with the increasing potential, at the same time a new broad band in the vicinity of 780 nm grows. Taking into account that the intensity of the band corresponding to the π–π* transition in the poly(thienylene) backbone is not reduced, it is reasonable to assume that the observed changes are associated with the oxidative doping of the tetraaniline side groups. In fact the π–π* transition band is slightly growing at the beginning of the oxidation process, which results from its overlap with the band at about 410 nm, usually observed for radical cations in oxidized aniline oligomers.35 Consistent with the cyclic voltammetry, above E = +0.30 V the oxidative doping of the main chain takes place as manifested by the decrease of the intensity of the band at about 400 nm and a quick growth of the doping induced band peaked at 780 nm.
Let us be reminded that at E = −0.20 V, in the acidified electrolyte, the side tetraaniline groups in P4 are in the fully reduced state whereas in the non-acidified one they remain semi-oxidized. This leads to different spectroelectrochemical behaviour of this copolymer in the presence of the DPP protonating agent (see Fig. 4d). The oxidation of the fully reduced side oligoaniline chains starts at relatively low potentials and results in a decrease of the intensity of the band at 300 nm with simultaneous growth of a very broad absorption peaked at about 1050 nm. These changes strongly indicate that the reduced neutral tetraaniline substituents are being transformed into the semi-oxidized protonated ones. Note that at E = +0.20 V which correspond to the open circuit potential the registered spectrum is almost the same as that of the as prepared P4 after its protonation with DPP. This means that the process of the reduction of the semi-oxidized tetraaniline side group is reversible, at least in the chemical sense. At higher potentials doping of the poly(thienylene) main chain starts as seen by a decrease of the band at 403 nm and a growth of the doping induced band in the vicinity of 800 nm. The latter grows to the potential of E = +0.50 V, then it starts to decrease. This can be taken as a spectroscopic sign of the oxidative degradation especially in view of the fact that at potentials exceeding the second anodic peak in the cyclic voltammogram (E > +0.60 V) this process is accelerated.
EPR spectroelectrochemistry
The oxidation of P1, P3 and P4 results either in spin carrying radical cations (polarons) or spinless dications (bipolarons). Possible co-existing species are presented in Scheme 4. EPR coupled to electrochemistry is the most convenient method to study their formation and possible inter-conversion during the oxidative doping.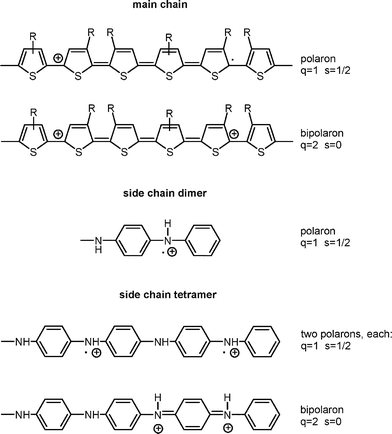 | ||
| Scheme 4 | ||
Fig. 5 shows the changes in the EPR magnetic susceptibility as a function of the electrode potential registered for P1. It should be noted that in all comparisons the potential of our pseudo-reference Ag electrode should be corrected by −0.30 V with respect to the Ag/Ag+ electrode used in other experiments, as shown by the determination of their potentials against the Fc–Fc+ couple. As expected in the neutral state P1 gives no EPR signal. At E = +0.88 V vs. Ag i.e. at +0.58 vs. Ag/Ag+ the spins appear and their number steadily increases with increasing potential in a relatively narrow potential range (up to +1.30 V). The observed behaviour is consistent with the shape of the cyclic voltammogram (Fig. 2a) which shows one narrow anodic peak, although at slightly lower potentials. The potentials measured during the EPR spectroelectrochemical experiment should however be treated with caution due to a very unfavourable geometrical conditions. It is clear that the oxidative doping gives rise to the formation of polarons. It should be noted here that this observation does not exclude the presence of bipolarons, since they are undetectable by EPR. The determination of both species requires the establishment of the spin–charge relationship which was not possible in the experimental conditions used, the main reasons being as follows: (i) extremely small quantities of the polymer were deposited on the Pt wire electrode which excluded the possibility of its mass determination by weighing. (ii) This mass could in principle be determined by coulometry, assuming an appropriate doping level at a given potential. However the size of the electrochemical cell had to be limited to the size of an EPR tube, moreover the quantity of the electrolyte had to be minimized, otherwise the EPR measurement could be severely perturbed. In these conditions, for extremely small quantities of the polymer, the Faraday yield for the doping process is never close to 100% which puts severe limits on the reliability of the spin–charge relation data.
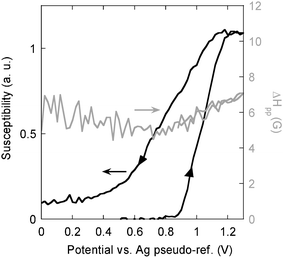 | ||
| Fig. 5 EPR susceptibility and EPR linewidth (ΔHpp) as a function of the electrode potential, measured during the electrochemical doping and dedoping of P1, electrolyte −0.1 M Bu4NBF4–acetonitrile solution. | ||
The fact that the susceptibility increases in a monotonic way seems to suggest that the recombination of polarons into bipolarons, if any, is not very efficient. Upon the reductive dedoping, the EPR susceptibility decreases, and a clear hysteresis is observed, consistent with the cyclic voltammetry results. If the doping process was totally reversible in the chemical sense, the magnetic susceptibility should have dropped to zero at the potentials corresponding to the total dedoping of the polymer. In reality, at low potentials, residual spins are present which constitute ca. 8% of the maximum number of spins created upon the oxidative doping. The presence of these residual, electrochemically inactive spins can be considered as the spectroscopic manifestation of the incomplete reversibility of the doping process. In Fig. 6, representative EPR lines registered at different potentials, both in the oxidation and in the reduction half-cycles, are compared. First we should note that all lines are strictly Lorentzian in shape, and their peak-to-peak width (ΔHpp) varies from 4.5 to 6 G, reaching its minimum in the middle of the reduction process. They are very similar to the corresponding lines observed upon positive charging of the other polythiophene derivatives.36,37
 | ||
| Fig. 6 EPR spectra of P1 registered at various potentials in the oxidation and reduction mode in 0.1 M Bu4NBF4–acetonitrile solution. | ||
Branching of the aniline tetramer to the poly(thienylene) main chain significantly changes the EPR electrochemical response of the polymer. In Fig. 7, the EPR susceptibility of P4 is plotted as a function of the electrode potential. First we note that even in the neutral state, a small but measurable number of spins exists, whose EPR parameters are characteristic of aniline tetramer radicals.38 The origin of these spins is not well understood at this time, but they always exist in the semi-oxidized form of polyaniline and its oligomers. Consistent with the cyclic voltammetry, the oxidative doping is manifested by the EPR susceptibility increase which starts at lower potentials as compared to the case of P1. In the oxidation part of the cycle no maximum of the spin susceptibility can be found. The evolution of the EPR line with increasing electrode potential is presented in Fig. 8. In the potential range covering the first anodic peak in the cyclic voltammogram, the EPR line is narrow with a peak-to-peak width of 2 G, which suggests the formation of radical cations (polarons) in the oligoaniline side groups. With increasing electrode potential, a broad component (ΔHpp = 5 G) of the spectrum appears and grows in intensity, constituting at the end of the oxidation the major contribution to the spin susceptibility. In the simplest interpretation, we ascribe this broad line to the formation of polarons in the conjugated main chain. Thus the EPR electrochemical response confirms the conclusions drawn from the cyclic voltammetry and the UV–vis–NIR spectroelectrochemical investigations. In the reduction part, the behaviour of P4 is different from that of P1. First, we notice a much larger hysteresis. Second, a clear maximum appears in the susceptibility vs. potential curve. We interpret it as an indirect proof of the existence of bipolarons together with polarons. If polarons were the sole charge storage configurations (one charge—one spin) a one electron reduction should inevitably lead to spinless species and as a consequence lower the susceptibility. To the contrary, one electron reduction of a bipolaron produces a spin-carrying polaron, which contributes to the total susceptibility. The observed maximum suggests therefore the following sequence of processes: bipolarons→polarons→neutral undoped. The entities being formed are shown in Scheme 4. Finally, we should note that in the case of P4, the oxidation–reduction cycle is much less reversible as compared to P1, since the number of residual spins accounts for ca. 30% of the total number of spins. Thus irreversibility is additionally confirmed by a growing number of residual spins, at the reduced neutral state, after each consecutive oxidation–reduction cycle.
 | ||
| Fig. 7 EPR susceptibility and EPR linewidth (ΔHpp) as a function of the electrode potential, measured during the electrochemical doping and dedoping of P4, electrolyte −0.1 M Bu4NBF4–acetonitrile solution. | ||
 | ||
| Fig. 8 EPR spectra of P4 registered at various potentials in the oxidation and reduction mode in 0.1 M Bu4NBF4–acetonitrile solution. | ||
The acidification of the electrolyte by DPP modifies the electrochemical response of P4 as probed by the EPR. Before the discussion of the electrochemical spin response of P4 it is instructive to discuss the EPR properties of aniline tetramer in the protonated form. In the “free” semi-oxidized tetramer the protonation causes a significant increase in the number of spins, as determined by EPR at room temperature, from ca. 10−5 spins per tetramer unit in the nonprotonated form to ca. 3 × 10−1 per tetramer unit in the protonated one.38 This introduction of unpaired spins, in the protonated state, occurs via the well known mechanism of charge rearrangement leading to the semi-quinone radical structure (see Scheme 4). Another peculiar feature of protonated polyaniline and its oligomers is a significant broadening of their EPR line as a result of interactions with paramagnetic species present in the environment or in the medium in which the sample is tested.39 Typical chemical species inducing the broadening of the oligoaniline EPR line are oxygen molecules, cations of transition metals and others. The determination of the intrinsic linewidth requires in all these cases the removal of all sources of paramagnetism, for example removal of oxygen by extending pumping in a vacuum line.
All these specific features of the semi-oxidized, protonated state of aniline tetramer are retained upon its grafting to the poly(thienylene) backbone. As expected, in P4 at E = 0.02 V i.e. at the potential where the main chain is in the reduced, neutral state and the side oligoaniline group in the semi-oxidized protonated state a strong EPR signal is recorded consistent with the protonation induced formation of polarons. As seen in Fig. 9 the EPR line is inhomogeneusly broadened due to the interactions with paramagnetic oxygen dissolved in the electrolyte. This observation indicates a very strong sensitivity of the recorded EPR line with respect to oxygen since the electrolyte was deoxygenated prior to the measurement by argon bubbling. However complete removal of the oxygen absorbed in the polymer layer is not possible even after a long series of freeze-pump deoxygenations.
 | ||
| Fig. 9 EPR spectra of P4 registered at various potentials in the oxidation and reduction mode in the acidified electrolyte (0.1 M Bu4NBF4 + 0.1 M diphenyl phosphate–acetonitrile solution). | ||
The presence of the “background” spins, which in addition dominate over the electrochemically created ones, makes the electrochemical-EPR response more complex. Fig. 10a shows the variation of the EPR susceptibility of P4 with the electrode potential. A clear maximum is observed at E = +0.65 V. Two processes should be taken into consideration in the discussion of the origin of this maximum. The first process involves the formation of the main chain polarons followed by their recombination to bipolarons. This explanation requires the assumption than the oxidation mechanism of the main chain in P4 in acidified electrolytes is different from that of P1 and P4 in non-acidified ones. There is no simple justification for such a change in behaviour. We propose therefore another explanation. It is known that protonated semi-oxidized polyaniline (or its oligomers), upon further oxidation to the fully oxidized (pernigraniline) state, undergoes deprotonation which leads to a neutral and spinless product. The observed susceptibility decrease may therefore originate from the oxidation induced deprotonation. Spectroscopic differentiation between both proposed processes is not easy because the main chain poly(thienylene) radicals and the side chain ones interacting with the paramagnetic environment, show similar g and ΔHpp. As a result only an inhomogeneously broadened line is observed.
 | ||
| Fig. 10 (a) EPR susceptibility and EPR linewidth (ΔHpp) as a function of the electrode potential, measured during the electrochemical doping and dedoping of P4 in acidified electrolyte (0.1 M Bu4NBF4 + 0.1 M diphenyl phosphate–acetonitrile solution) (b) time evolution of the EPR susceptibility at constant electrode potential of E = +0.80 V vs. Ag. | ||
In the reductive part of the cycle the EPR susceptibility steadily increases. The initial increase is understandable since it should involve the contribution of two processes to the increase in the number of spins: (i) one electron reduction of a spinless bipolaron which gives a spin carrying polaron and (ii) spin creating reprotonation of the side chain accompanying their reduction from the fully oxidized state to the semi-oxidized one. However at lower potentials the susceptibility should decrease because of the one-electron reduction of spin carrying polarons, both in the main chain and in the side groups, to the spinless neutral state. We observe the opposite i.e. the EPR susceptibility monotonically increases upon the reduction which may mean that the inhibition of the reduction to the neutral state may be of kinetic origin. This is probably the case since during the second oxidation cycle if the oxidation is stopped at E = +0.80 V and the sample is kept at this constant potential, the EPR susceptibility decreases to the value measured at the same potential in the first oxidative cycle within 30 min (see Fig. 10 b).
Raman spectroelectrochemistry
The evolution of the Raman spectra of P1, with increasing electrode potential, should reflect its oxidative doping. P1 gives a clear Raman spectroelectrochemical response which, as expected from its chemical constitution, much more resembles the behaviour of HH–TT coupled poly(4,4′-dialkyl-2,2′-bithiophene)s than that of HT–HT coupled poly(3-alkylthiophene)s.40In Fig. 11 Raman spectra of P1, recorded for increasing electrode potentials are collected. Since P1, in its undoped state, is out of resonance with the infrared excitation line (λexc = 1064 nm) as judged from its UV–vis–NIR spectrum (Fig. 4a), it is practically not possible to register its Raman spectrum at potentials below the onset of the first anodic peak. For the undoped polymer only one, very weak band can be distinguished at 1490 cm−1i.e. in the spectral range where either Cα–Cβ symmetric or antisymmetric stretching vibrations are expected.41 Consistent with UV–vis–NIR spectroelectrochemistry (Fig. 4a), the resonance conditions suddenly improve at E = +0.43 V where a significant increase in the absorbance value is observed at λ = 1064 nm. As a consequence a clear, good quality Raman spectrum can be registered. The observed bands are characteristic of the doped (oxidized state). In particular two principal, strongly overlapping bands at 1395 cm−1 and at 1381 cm−1 can be ascribed to the Cα–Cβ stretching deformations which are characteristic of the presence of head to head (HH) couplings in the polymer chain.40 In the HT–HT coupled polymers only one band is present in the Raman spectrum.41 These lines are red shifted with respect to the corresponding lines in the neutral polymer which is a consequence of the doping induced force constant weakening.41 The bands originating from the Cα–Cβ stretching deformations are accompanied by a doublet at 1227 cm−1 and at 1204 cm−1 assigned to the Cα–Cα′ (inter-ring) stretchings,41 which become more pronounced upon doping. Clear lines in the spectral range 690–750 cm−1 originate from the C–S deformations in the thiophene ring. Note that the line originating from the Cβ–Cβ stretchings is present only as a shoulder of the line peaked at 1395 cm−1 (Cα–Cβ stretchings). As soon as it appears, the Raman spectrum of P1 does not change its shape up to E = +0.60 V. This means that we detect only the doped segments being in resonance with the excitation line (λ = 1064 nm). Above E = +0.60 V the Raman lines start to significantly broaden which is a clear indication of the increasing oxidative degradation of the polymer which usually follows the oxidative doping at higher potentials. Once again it should be underlined that the Raman spectroelectrochemical results are very consistent with those obtained by cyclic voltammetry and UV–vis spectroelectrochemistry.
 | ||
| Fig. 11 Raman spectra of the precursor polymer P1 deposited on a Pt electrode, registered for increasing electrode potentials, λexc = 1064 nm; potential vs. Ag/0.1 M Ag+ reference; 0.1 M Bu4NBF4–acetonitrile solution. | ||
Grafting of tetraaniline groups to the precursor polymer, i.e. transforming P1 into P4 strongly alters the resonance conditions. The new substituents are electroactive chromophores whose Raman response is additionally sensitive to the presence of an appropriate protonating agent in the electrolyte. We first examine the case with no protonating agent in the electrolyte solution. At the open circuit potential (E = −0.30 V) P4 does not give any measurable Raman spectrum. In fact no spectrum can be registered in the potential range −0.20 V < E < +0.05 V (see Fig. 12). This is not unexpected since, as shown by UV–vis spectroelectrochemistry, at these potentials, neither the main polythiophene chain chromophore nor the side oligoaniline one contribute to the absorbance at 1064 nm and all subunits constituting P4 are out of resonance for this excitation line. Measurable Raman spectrum appears at E = +0.05 V giving lines at 1179, 1385, 1510, 1602 and 1626 cm−1. All these line are characteristic of the oligoaniline substituent.42 The lack of the bands attributable to the polythiophene backbone at this potential range corroborates the conclusion, drawn from the UV–vis spectroelectrochemistry results, that the oligoaniline side chains are oxidatively doped first. The presence of two Raman lines in the spectral range characteristic of quinone and benzene vibrations (1595 and 1623 cm−1) seems to indicate the co-existence of both types of rings in the doped polymer. This means that the bipolaronic form presented in Scheme 4 is in stronger resonance than the polaronic one since the latter should give rise to one line at an intermediate position.42 To the contrary, the EPR spectroelectrochemistry shows the creation of spins in the potential range characteristic of the side chain oxidation, unequivocally indicating the presence of the spin carrying polaronic form (Scheme 4). Thus both forms (polaronic and bipolaronic) must co-exist in the polymer being selectively detected by the applied techniques (EPR and resonant Raman).
 | ||
| Fig. 12 Raman spectra of the hybrid polymer P4 deposited on a Pt electrode, registered for increasing electrode potentials, λexc = 1064 nm; potential vs. Ag/0.1 M Ag+ reference; 0.1 M Bu4NBF4–acetonitrile solution. | ||
In the potential range from E = +0.05 V to E = +0.50 V the shape of the Raman spectrum does not change which means that in each case the same doped side groups are detected because of the resonant enhancement. Above E = +0.50 V, the intensity of the lines originating from the tetraaniline side groups decreases and the lines ascribed to the oxidation of the polythiophene main chain appear and grow in intensity. At the highest potentials studied only the bands characteristic of the oxidized polythiophene main chain are present, however significant broadening of these lines seems to suggest that the polymer is partially degraded. We have observed that at E = +0.90 V the spectrum of the precursor polymer P1 and that of P4 are almost identical.
Raman spectroelectrochemical behaviour of P4 in the acidified electrolyte is different. First, it should be noted that at the open circuit potential, the polymer gives a clear Raman spectrum characteristic of the protonated state of semi-oxidized oligoaniline side chains. Again, the co-existence of two lines at 1595 and 1623 cm−1 seems to indicate that the bipolaron form (Scheme 4) is in resonance (Fig. 13). The protonation of the oligoaniline side chains gives rise to the resonant Raman enhancement since it involves a significant increase of the absorbance at 1064 nm—the wavelength of the used excitation line. The reduction of the “as prepared” polymer at E = −0.20 V results in a total disappearance of its Raman spectrum, consistent with a decrease of the absorbance at 1064 nm (see Fig. 3). Evidently, at this potential, none of the polymer segments are in resonance with the infrared excitation line. Upon oxidation of this totally reduced form of P4, the spectrum reappears at E = +0.10 V giving lines characteristic of the protonated semi-oxidized form of the oligoaniline side groups. At E = +0.20 V the spectrum is identical to that registered at the open circuit potential proving that in the acidified electrolyte the redox processes in the potential range of the first redox couple are reversible, at least in the chemical sense. This is not unexpected since the semioxidized, protonated form of the aniline tetramer has, in its cationic part, the same stoichiometry as the fully reduced form. As a consequence the switching between these forms involves only the electron transfer.
 | ||
| Fig. 13 Raman spectra of the hybrid polymer P4 deposited on a Pt electrode, registered at Eoc (“as prepared” polymer), at E = −0.2 V (reduced polymer) and then for increasing electrode potentials up to +0.80 V, λexc = 1064 nm; potential vs. Ag/0.1 M Ag+ reference; 0.1 M Bu4NBF4–0.1 M DPP–acetonitrile solution. | ||
In the potential range of +0.10 V < E < +0.50 V the lines characteristic of the protonated semi-oxidized form of the tetramer side group grow in intensity as a result of improving resonance conditions for this form. Evidently the continuous increase of the absorbance at 1064 nm, registered for increasing electrode potentials is caused, in this case, by growing content of the protonated side-group tetramer. Above E = +0.50 V the resonance conditions worsen and the intensity of the observed Raman lines diminishes. It should be stressed that the lines originating from the polythiophene main chain remain “invisible” for the Raman spectroscopy throughout the whole potential range studied. At the highest potentials spectra of a very poor quality, with no distinguishable Raman lines, are registered. This can be considered as a spectroscopic manifestation of the polymer degradation. Evidently, for potentials exceeding the maximum of the second anodic peak the oxidative degradation of P4 is accelerated in acidified electrolytes.
At the end, it should be stressed that all three (UV–vis, EPR and Raman) spectroelectrochemical techniques together with cyclic voltammetry give a very clear and consistent response concerning the electrochemical oxidative doping of the precursor polymer P1 and its oligoaniline functionalized derivatives (P3 and P4).
Conclusions
To summarize, we have developed an efficient method of the preparation of alternate copolymers of dialkylbithiophenes and oligoanilinethiophenes in which the thiophene rings are head to head coupled. These solution processable polymers show very interesting electrochemical behaviour associated with two types of electroactive groups (side and main chains).In particular, their electrochemical doping is sequential which means that at lower electrode potentials they can be selectively doped in their oligoaniline side chains whereas at higher potentials the doping is global i.e. involves both the side chains and the polythienylene main chain. This conclusion is drawn from a careful analysis of a consistent evolution of UV–vis, Raman and EPR spectra registered for increasing electrode potentials and from the correlation of these changes with the observed voltammetric features. Spectroelectrochemical investigations show additionally that the redox processes in the electroactive side chains are facilitated by protonation–deprotonation phenomena occurring in acidified electrolytes.
Acknowledgements
Some of the authors (K.B., A.M., R.P., M.Z.) acknowledge financial support by Warsaw University of Technology.References
- R. D. McCullough, Adv. Mater., 1998, 10, 93 CrossRef CAS.
- M. Besbes, G. Trippé, E. Levillain, M. Mazari, F. Le Derf, I. F. Perepichka, A. Derdour, A. Gorgues, M. Sallé and J. Roncali, Adv. Mater., 2001, 13, 1249 CrossRef CAS and references therein.
- J. Lowe and S. Holdcroft, Macromolecules, 1995, 28, 4608 CrossRef CAS.
- N. Miyaura, Topics in Current Chemistry: Cross-Coupling Reactions, Vol. 219, Springer, Berlin, 2002 Search PubMed.
- Y. Li, G. Vamvounis and S. Holdcroft, Macromolecules, 2002, 35, 6900 CrossRef CAS.
- M. Lanzi, L. Paganin, P. Costa-Bizzarri, C. Della-Casa and A. Fraleoni, Macromol. Rapid Commun., 2002, 23, 630 CrossRef CAS.
- M. Lanzi, L. Paganin and P. Costa-Bizzarri, Eur. Polym. J., 2004, 40, 2117 CrossRef CAS.
- G. Trippé, F. Le Derf, J. Lyskawa, M. Mazari, J. Roncali, A. Gorgues, E. Levillain and M. Sallé, Chem. Eur. J., 2004, 10, 6497 CrossRef CAS.
- P. Bäuerle, M. Hiller, S. Scheib, M. Sokolowski and E. Umbach, Adv. Mater., 1996, 8, 214.
- A. Iraqi, J. A. Crayston and J. C. Walton, J. Mater. Chem., 1998, 8, 31 RSC.
- G. Li, G. Koßmehl, W. Hunnius, H. Zhu, W. Kautek, W. Plietch, J. Melsheimer and K. Doblhofer, Polymer, 2000, 41, 423 CrossRef CAS.
- L. Zhai, R. L. Pilston, K. L. Zaiger, K. K. Stokes and R. D. McCullough, Macromolecules, 2003, 36, 61 CrossRef CAS.
- G. Zotti, S. Zecchin, G. Schiavon, B. Vercelli and A. Berlin, Electrochim. Acta, 2005, 50, 1469 CrossRef CAS.
- B. Dufour, P. Rannou, J. P. Travers, A. Pron, M Zagorska, G. Korc, I. Kulszewicz-Bajer, S. Quillard and S. Lefrant, Macromolecules, 2002, 35, 6112 CrossRef CAS.
- K. Buga, K. Kępczyńska, I. Kulszewicz-Bajer, M. Zagórska, R. Demadrille, A. Pron, S. Quillard and S. Lefrant, Macromolecules, 2004, 37, 769 CrossRef CAS.
- K. Buga, A. Majkowska, R. Pokrop, M. Zagórska, D. Djurado, A. Pron, J.-L. Oddou and S. Lefrant, Chem. Mater., 2005, 17, 5754 CrossRef CAS.
- R. Demadrille, B. Divisia-Blohorn, M. Zagorska, S. Quillard, S. Lefrant and A. Pron, Electrochim. Acta, 2005, 50, 1597 CrossRef CAS.
- D. K. Grant and D. L. Officer, Synth. Met., 2005, 154, 93 CrossRef CAS.
- S. Gambihr, K. Wagner and D. Officer, Synth. Met., 2005, 154, 117 CrossRef.
- P. Costa-Bizzarri, F. Andreani, C. Della Casa, M. Lanzi and E. Salatelli, Synth. Met., 1995, 75, 141 CrossRef CAS.
- B.-S. Kim, L. Chen, J. Gong and Y. Osada, Macromolecules, 1999, 32, 3964 CrossRef CAS.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522 CrossRef.
- T. Kirschbaum, R. Azumi, E. Mena-Osteritz and P. Bauerle, New J. Chem., 1999, 23, 241 RSC.
- J. Pei, J. Ni, X.-H. Zhou, X.-Y. Cao and Y.-H. Lai, J. Org. Chem., 2002, 67, 4924 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- S. Gronowitz and D. Peters, Hetercycles, 1990, 30, 645 Search PubMed.
- G. E. Collis, A. K. Burrell, S. M. Scott and D. L. Officer, J. Org. Chem., 2003, 68, 8974 CrossRef CAS.
- R. Demadrille, P. Rannou, J. Bleuse, J. L. Oddou, A. Pron and M. Zagorska, Macromolecules, 2003, 36, 7045 CrossRef CAS.
- J. S. Liu and R. D. McCullough, Macromolecules, 2002, 35, 9882 CrossRef CAS.
- P. Barta, P. Dannetun, S. Stafstrom, M. Zagorska and A. Pron, J. Chem. Phys., 1994, 100, 1731 CrossRef CAS.
- R. P. McCall, J. M. Ginder, J. M. Leng, H. J. Ye, S. K. Manohar, J. G. Masters, G. E. Asturias, A. G. MacDiarmid and A. J. Epstein, Phys. Rev. B: Condens. Matter, 1990, 41, 5202 CrossRef CAS.
- Y. Xia, J. M. Wiesinger and A. G. MacDiarmid, Chem. Mater., 1995, 7, 443 CrossRef CAS.
- M. Lapkowski, M. Zagorska, I. Kulszewicz-Bajer, K. Koziel and A. Pron, J. Electroanal. Chem., 1991, 310, 57 CrossRef CAS.
- C. H. McCoy, I. Lorkovic and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 6934 CrossRef CAS.
- I. Rozalska, P. Kulyk and I. Kulszewicz-Bajer, New J. Chem., 2004, 28, 1235 RSC.
- A. Zykwinska, W. Domagala, B. Pilawa and M. Lapkowski, Electrochim. Acta, 2005, 50, 1625 CrossRef CAS.
- A. Cravino, H. Neugebauer, S. Luzzati, M. Catellani, A. Petr, L. Dunsch and N. S. Sariciftci, J. Phys. Chem. B, 2002, 106, 3583 CrossRef CAS.
- R. Payerene, PhD Thesis, Joseph Fourier University, Grenoble, 2005 Search PubMed.
- E. Houze and M. Nechtschein, Phys. Rev. B: Condens. Matter, 1996, 53, 14309 CrossRef CAS.
- A. Pron, M. Lapkowski, M. Zagorska, J. Glowczyk-Zubek, G. Louarn and S. Lefrant, in Photoactive Organic Materials, Science and Application, ed. F. Kajzar, V. M. Agranovich, C. Y.-C. Lee, NATO ASI Series, 3. High Technology, 1996, 9, 361 Search PubMed.
- G. Louarn, M. Trznadel, J. P. Buisson, J. Laska, A. Pron, M. Lapkowski and S. Lefrant, J. Phys. Chem., 1996, 100, 12532 CrossRef CAS.
- M. Lapkowski, K. Berrada, S. Quillard, G. Louarn, S. Lefrant and A. Pron, Macromolecules, 1995, 28, 1233 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: synthesis and characterization of monomer (3), 1H NMR of all studied polymers, the P3 cyclic voltammograms dependence on potential scan rate. See DOI: 10.1039/b516677m |
| This journal is © The Royal Society of Chemistry 2006 |