Prediction of thermodynamic stability and electronic structure of novel ternary lanthanide hydrides†
Tomasz
Jaroń
ab,
Wojciech
Grochala
*ac and
Roald
Hoffmann
*b
aDepartment of Chemistry, University of Warsaw, Pasteur 1, 02-093 Warsaw, Poland. E-mail: wg22@cornell.edu; Fax: +48 22 8225996 ext. 276; Tel: 48 22 8220211
bDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853-1301, USA. E-mail: rh34@cornell.edu; Fax: +1 607 2555707; Tel: +1 607 2553419
cInterdisciplinary Center for Mathematical and Computational Modeling, University of Warsaw, Pawińskiego 5a, 02-106 Warsaw, Poland
First published on 3rd January 2006
Abstract
We theoretically examine four hypothetical ternary lanthanide hydrides, CsLnIIH3 and Cs2LnIIH4, where LnII = Yb, Tm. We optimize their crystal unit cells in the BaTiO3 and K2NiF4 structures, respectively, and compute their electronic band structures. Our calculations indicate that the novel hydrides should be unstable with respect to decomposition into binaries (CsH and LnIIH2); ternaries would form only under elevated pressure (>7–22 GPa). We predict that significant perturbation of the electronic and magnetic properties of CsYbIIH3 and Cs2YbIIH4 will take place via a progressive exchange of f14 YbII for f13 TmII, while promoting magnetic ordering and valence fluctuations. Analysis of the phonon dispersion for these hydrides suggests that metallic forms of doped CsLnIIH3 and Cs2LnIIH4 would exhibit substantially high Debye temperatures of ∼1800 K. This is likely to prompt moderate-TC superconductivity in these as yet unknown materials.
Introduction
Among lanthanide metals, the divalent state is favored predominantly for Yb and Eu, due to the stability of—respectively—filled (f14) and half-filled (f7) f shell. Significantly lesser stability is noticed for the elements preceding Yb and Eu in the periodic table, namely Tm and Sm, respectively.1 This is attested to by the standard redox potentials for the MIII/MII redox pairs (E0 = −1.05 V for Yb, while −2.30 V for Tm; −0.35 V for Eu, while −1.55 V for Sm).2 As a consequence, the nearly stoichiometric trihydrides of trivalent Tm and Sm are known, while genuine YbH3 and EuH3 have never been synthesized. For Yb only the mixed-valence YbII(YbIIIH4)2 has been obtained so far,3 while for Eu, EuH1.95 is a limiting composition, in agreement with the ranking of the E0 values and other thermodynamic arguments.4YbII and EuII, the most prominent representatives of stable divalent lanthanide cations, are often compared to closed shell CaII and SrII, respectively, in terms of ionic radius.5 A nice series of isomorphic compounds of YbII and CaII, and those of EuII and SrII, are known; examples include YbBeF4 and CaBeF4, EuLiF3 and SrLiF3, and—among the hydrides—Yb4Mg3H146 and Yb4Mg4Co3H197 (and their Ca analogues), Eu6Mg7H268 and inverse perovskite EuLiH3 (and their Sr analogues). In all these compounds, binary components of divalent Yb and Eu (i.e., EuF2, YbH2etc.) formally serve as Lewis bases, while donating their ligands to the stronger and smaller Lewis acids (MgII, BeII).
TmII (ionic radius, R, of 1.17 Å in the octahedral environment), an electron-deficient analogue of YbII, is virtually isomorphic with YbII (R = 1.16 Å)9 and with CaII (R = 1.14 Å). However, because of the enormous reducing properties of TmII,10 its compounds are scarce, and very difficult to preserve in the presence of traces of O2 or humidity.
Prompted by the existence of MICaX3 and MI2CaX4, where MI = Cs, Rb and X = H or F,11 of ternary lanthanide salts such as the perovskites CsYbF3,12 CsYbI3 and RbTmI3,13 and driven by the fluoride-hydride analogy,14 we examine theoretically and attempt to predict the possible existence and selected properties of CsLnIIH3 and Cs2LnIIH4 where LnII = Yb, Tm. In these hypothetical compounds, and in contrast to all known ternary hydrides containing divalent lanthanide cations, LnH2 would formally serve as a Lewis acid while accepting hydride anion from the very strong base (CsH). Here, our interest in ternary hydrides of divalent lanthanides has origin in (i) anticipated involvement of f orbitals in bonding, i.e. a mixing of LnII (4f) and H (1s) orbitals (moderate ‘covalence’), (ii) large values of electronic density of 4f states in the vicinity of the Fermi level, and (iii) concomitant consequences of hole- and electron-doping for electronic and magnetic properties of the stoichiometric compounds of LnII, and possible promotion of superconductivity.
Methods of calculations
Our computations were based on density functional theory and used a generalized gradient approximation (GGA) with ultrasoft Vanderbilt-type pseudopotentials, as implemented in the CASTEP15 code. For each structure, we relaxed the unit cell constants and atomic positions (whenever possible), and also determined the stress tensor. The electronic density of states, band structure and electronic density within a desired energy window were subsequently calculated for the optimized unit cell. The k-point grids for zone sampling were generated automatically via the Monkhorst–Pack scheme. The k-point sampling used by CASTEP was 4 × 4 × 4 (structures of CsLnH3), and 4 × 4 × 2 (Cs2LnH4). We used a 450 eV cutoff for the kinetic energy of the plane waves and an SCF tolerance of 2 × 10−6 eV per atom. In our calculations we used plane waves originating from an expansion of four valence and sub-valence orbitals for Cs (5s2, 5p6, 6s2, 6p6), two orbitals for each H (1s, 2s) and six orbitals for Yb and Tm (5s2, 5p6, 4f14, 6s2, 6p2, 5d10). This gives a total of 31 bands for CsYbH3 (Z = 1); 18 of these are occupied, 13 are unoccupied. No other atomic functions (in particular no polarization functions on H, 2p) are embedded in the CASTEP package for these atoms.For compounds of TmII (an open-shell f13 configuration with large magnetic moment) we have always done spin-polarized calculations (SP). Calculations without spin polarization (‘metallic’ or ‘antiferromagnetic’ polymorph) always yielded a larger energy per unit cell (by about 0.5 eV per Tm atom) than for those with polarized spins (‘ferromagnetic’ polymorph). At the same time, calculations without spin polarization typically result in minor changes in the unit cell vectors (for example, compare results for Cs2TmH4: SP a = b = 4.601 Å, c = 15.696 Å, no SP a = b = 4.616 Å, c = 15.613 Å).
We have also performed supplementary VASP16 calculations for several Yb- and Tm-containing compounds (see the electronic supplementary information (ESI)†). Here we could use dense k-point sampling of 11 × 11 × 11 (CsLnH3), and 11 × 11 × 6 (Cs2LnH4). We tested two types of pseudopotentials: those which explicitly involve f electrons on Ln atoms (PBA set) and those which do not; it turned out that optimizations with VASP (pseudopotentials without f electrons) and CASTEP (pseudopotentials with f electrons) yielded only minor differences in unit cell vectors for CsYbH3 and Cs2YbH4.
For phonon dispersion and phonon DOS calculations we have used the PHONON17 code as implemented in the MEDEA package. PHONON uses VASP-preoptimized unit cells in order to determine vibrations of the crystal lattice. To speed up the time-demanding frequency calculations we used norm-conserving pseudopotentials which do not explicitly involve f electrons on Ln atoms.
Results and discussion
Crystal structures
Due to the CaII–LnII (Ln = Yb, Tm) isomorphism mentioned above, one may expect that CsLnIIH3 and Cs2LnIIH4 will adopt, respectively, the CsICaH3 (perovskite) and CsI2CaH4 (K2NiF4) structures. In Table 1 we present the results of unit cell optimization for four novel ternary lanthanide hydrides, and compare them with the experimental and computational data for analogous calcium salts.11| Compound | CsYbH3 | CsTmH3 | CsCaH3 | CsCaD3 exp. | Cs2YbH4 | Cs2TmH4 | Cs2CaH4 | Cs2CaD4 exp. |
|---|---|---|---|---|---|---|---|---|
| a = b/Å | 4.609 | 4.609 | 4.619 | 4.617 | 4.609 | 4.601 | 4.615 | 4.597 |
| c/Å | 4.609 | 4.609 | 4.619 | 4.617 | 15.531 (0.148) | 15.696 (0.146) | 15.558 (0.149) | 15.528 (0.149) |
Our GGA-PBA calculations predict a cubic unit cell constant of 4.609 Å for CsYbH3 and 4.609 Å as well for CsTmH3; the deviation from the experimental value for CsCaD3 (4.617 Å) is very small (<0.01 Å) and slightly negative; in fact small positive deviations (ca. +0.03 to 0.04 Å) are expected based on comparison of the crystal radii (see the previous section). The difference between calculated and experimental cubic unit cell constants for CsCaD3 is as small as 0.002 Å.
Our result is entirely within the limitations of the computational method utilized (we note slightly positive deviations for smaller energy cutoffs). The calculated unit cell constant of 4.609 Å for CsYbH3 also matches well the experimental value of 4.61 Å for CsYbF3; indeed, the close hydride–fluoride analogy has been pointed out to apply for similar compounds.14 For CsTmH3, the environment of TmII is predicted to be that of an ideal octahedron; the Jahn–Teller (JT) distortion apparently does not influence the coordination shell of the f13 cation, similar to the crystal structure of CsTmCl3.18 This feature points to the relatively ionic character of Tm–H bonding.
For Cs2YbH4, slight deviations from regular octahedral coordination of YbII are predicted, with four equivalent longer equatorial Ln–H distances of 2.305 Å and two shorter apical distances of 2.301 Å. This effect is of course slightly larger for ferromagnetic (f13) Cs2TmH4 (equatorial 2.300 Å, apical 2.286 Å). An inverse JT effect is computed to be small for the inner transition metal f13 configuration of TmII (the ratio of apical to equatorial Tm–H distances is 0.994), in agreement with experimental data for similar compounds of TmII (the analogous ratio being 0.986 for KTmI3,19 and 1.006 for Cs2TmCl420).
The fractional z atomic coordinate for an apical H(0,0,z) atom (which quantitatively determines the distortion of the {LnH6} octahedron), has been predicted to be 0.146 for Cs2TmH4, via 0.148 for Cs2YbH4, up to 0.149 for Cs2CaH4, always close to the experimental value of 0.149 for Cs2CaD4.
The predicted unit cell vectors are very similar for hydrides of YbII and for isostructural compounds of TmII (identical for CsLnH3, and within 1% deviation for Cs2LnH4). This should facilitate formation of solid solutions for analogous compounds of these Ln metals, i.e., ease of electronic doping.
Thermodynamic stability
In Table 2 we present the calculated enthalpies of formation21 (at T = 0) of the ternary caesium lanthanide hydrides studied, with respect to formation from CsH and LnH2 (CsH and LnH2 have been optimized based on the experimental crystallographic data22).| Compound | CsYbH3 | CsTmH3 | Cs2YbH4 | Cs2TmH4 |
|---|---|---|---|---|
| ΔH/eV per molecule | 0.27 | 0.55 | 0.19 | 0.47 |
| ΔV/Å3 per molecule | −6.02 | −5.30 | −5.66 | −3.85 |
| p/GPa | 7.3 | 16.7 | 5.3 | 19.7 |
Ternary Tm and Yb hydrides are predicted to be thermodynamically unstable with respect to decomposition to binary hydrides, and it is not expected that vibrational zero-point correction and entropy effects—typically small for solid reagents—will easily reverse this trend. Diamagnetic forms of TmII compounds are computed to have higher electronic energy than their magnetically ordered (ferromagnetic) polymorphs, in agreement with the strong magnetism of unpaired f electrons. In the following sections we will discuss exclusively ferromagnetic compounds of TmII.
The enthalpy of the reactions of synthesis of ternary hydrides is positive for all ternaries studied, while the associated volume change is computed to be negative (Table 2). This suggests that hydrides might form from binaries at elevated pressure. A rough estimate of the formation pressure can be obtained from the “common tangent” method, as a ratio of reaction enthalpy to the volume change. The estimated formation pressures—listed in Table 2—range from 5 to 20 GPa.23
Our conclusion is that ternary hydrides of TmII and YbII could be synthesized at increased pressures, and at relatively low temperatures up to 200 °C (to avoid decomposition of fragile CsH) and with rather small H2 overpressure (to avoid formation of compounds of trivalent lanthanides, especially for compounds of divalent Tm, strong reducing agents). Some of these ternary phases might be quenchable upon decompression to 1 atm.
Electronic structure
Let us now analyze the electronic structure of CsLnH3 and Cs2LnH4.The band structure of Cs/Yb(Tm) hydrides (see ESI†) consists of: (i) very narrow filled subvalence bands originating from 5s2 (at about −52 eV) and 5p6 (at about −24 eV) states of Yb(Tm), and from 5s2 (at about −23 eV) and 5p6 (at about −10 eV) states of Cs, (ii) a filled ‘hydride band’ at −6 to −2.5 eV, (iii) a very narrow 4f valence band and (iv) a very broad set of bands constituting the conducting band. In the analysis to come we will omit the sub-valence bands, i.e. set (i).
The CASTEP software does not allow direct atomic identification of a given segment of the density of states, only decomposition by angular momentum type—s, p, d, or f. Nevertheless an atomic assignment can be made, both by the energy range in which the density is found, and also by computing the disposition in space of the states in a given energy interval.
The ‘hydride band’ (ii) is visibly split from the valence band (iii) for all ternary hydrides studied (by some 3 eV) and it is composed predominantly of H (1s) states (Fig. 1), with a little contribution from lanthanide s, p and d states; in turn, the valence 4f band contains very little contribution from H (1s) states, which might point to an ionic formulation of the hydrides studied, to be written formally as CsILnII(H−)3 and (CsI)2LnII(H−)4. The ‘hydride band’ is usually split in several separate bands, some of which show significant dispersion (up to 3 eV). This cannot be due to direct H⋯H interactions, as the hydrides are far apart (usually at >3.2 Å). The band width in the hydride band is due to interactions between H ions and lanthanide and alkali centers; the title hydrides are not as ionic as one might think.
 | ||
| Fig. 1 Band structure and DOS for CsYbH3 (upper left), CsTmH3 (upper right), Cs2YbH4 (lower left) and Cs2TmH4 (lower right). Focus is on hydride, f and conduction bands. | ||
The set of bands (iii) originating from 4f states either gives rise to a single and large peak in the DOS (as for diamagnetic f14 YbII) or is split equally into two bands separated by some 2 eV (as for ferromagnetic f13 TmII). This description of f electrons in these phases is unsatisfactory; unfortunately it is common to all first-principles calculations within the independent electron approximation.24,25
The broad conduction band, or rather, the set of interpenetrating bands (with a collective width of up to 9.5 eV for CsLnIIH3 and of only 3 eV for Cs2LnIIH4) is built of s, p and d states, predominantly coming from Yb, but in some part also from the alkali metal. For Yb hydrides, the bottom of the (formally empty) conduction band is remarkably close (0.25–0.5 eV) to the uppermost part of the (formally filled) valence band. For Tm compounds, however, the broad conduction band penetrates through (and for CsTmH3 even 0.5 eV below) the narrow 4f band, thus closing the direct band gap.
The electronic density at the Fermi level consists mainly of rather localized 4f electrons (Fig. 2) with no contribution from H 1s states.
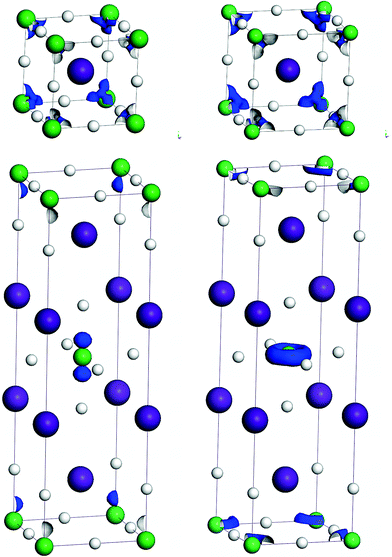 | ||
| Fig. 2 Electronic density (in blue) at the Fermi level (EF ± 0.03 eV) for CsYbH3 (upper left), CsTmH3 (upper right), Cs2YbH4 (lower left) and Cs2TmH4 (lower right). Density isovalue 0.012 e Å−3. | ||
Phonon density of states
Prompted by our continuing search for novel superconducting materials (with particular emphasis on fluorides and hydrides),26 and by an unprecedented dynamic scale of all H-containing solids, we have investigated the phonon dispersion and phonon density of states for CsYbIIH3 (Fig. 3).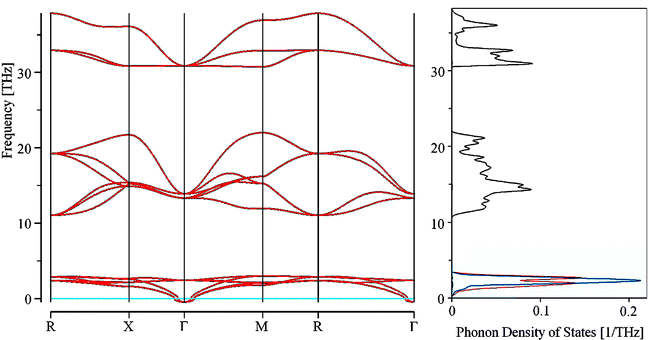 | ||
| Fig. 3 Phonon band structure (left) and phonon DOS (right) for CsYbH3; PHONON/VASP calculation. H's contribution to phonon DOS is in black, Yb's one in red, Cs's one in blue. | ||
The unit cell of CsYbH3 contains 5 atoms, so there are 15 degrees of freedom: 12 vibrational and 3 translational ones. Three phonon bands are seen in the highest frequency range of the spectrum (30.9–37.7 THz, 128–156 meV, 1030–1260 cm−1).27 They are degenerate at the zone center (recollect, the cell is cubic), and correspond to the Yb–H stretching modes. Six other modes are seen in the moderate-frequency range (11.0–21.9 THz, 46–90 meV, 370–730 cm−1). These are the Ln–H–Ln bending modes, which also show significant amplitude of H motions. The last set of three phonons is centered at low frequency (3.07 THz, 13 meV, 120 cm−1). These are deformational modes of the lattice, with predominant contribution from heavy elements (Yb, Cs). The remaining three lowest-frequency motions correspond to translations of the entire lattice; their calculated wavenumbers (at the zone center, Γ) are close to zero within narrow error margin, as expected. The dispersion of Ln–H stretching modes is significant (∼7 THz, 29 meV, 230 cm−1); that of Ln–H bending modes is even larger (∼10.8 THz, 45 meV, 360 cm−1).
The phonon density of states is composed of a single narrow and intense peak originating from the lowest-frequency phonons; a low dispersion, broad band coming from moderate-frequency phonons; and a double peak of moderate intensity marking the highest energy range of the spectrum.28
In the first section we have considered the ‘static’ crystal structure of lanthanide hydrides, and we have pointed to minor structural distortions predicted for the Cs2LnIIH4 compounds. We think that dynamic inversion of the Jahn–Teller effect (a set of local two short and four long Ln–H bonds transforms into two long and four short bonds) is undoubtedly within vibrational amplitude of H atoms, at least within the Born–Oppenheimer approximation. Interestingly, such fluctuating transformation of a JT species should decrease the anisotropy of electron density around the lanthanide cation, in particular for compounds of TmII.
Superconductivity in hydrides and valence fluctuations. Why is TC for hydrides so low?
As one knows for many other systems, moderate and high-temperature superconductivity arises in the vicinity of the metal-to-insulator transition.29 For mixed-valence systems, motions of ligands bridging two metal centers seem to be of crucial importance for phonon-driven superconductivity. Superconductivity has been detected for several hydride materials (for example Th4H15), yet despite the presence of a light H atom, the values of the transition temperature (TC) are remarkably small (not exceeding 20 K). In addition, the isotope effect on the value of TC is opposite to the predictions of the classical BCS theory30 for Pd hydride, the best studied hydride superconductor.31In a simplistic chemical picture, PdHx≈1.0 is formally a mixed valence system, Pd0PdIIH−12.32 A molecular mixed-valence systems can be either localized, delocalized (i.e., intermediate valence), or an intermediate case between the two.33 These three classes of molecular mixed-valence systems may naturally be compared to, respectively, insulating, metallic and superconducting mixed-valence solids.34 In this picture, the electronic wavefunction of a system (be it a solid or a molecule) is most affected by motions of nuclei when the vibrational zero-point energy (ZPE) of metal–ligand–metal stretching (or of the highest-energy optical phonon in solids) is close to the energy barrier for the electron transfer (EB) between the metal centers (Fig. 4).
 | ||
| Fig. 4 The double-well PES for a mixed-valence compound for three different zero-point vibrational energy situations: (A) a separated (localized) mixed valence compound; (B) a fluctuating valence species; (C) an intermediate (delocalized) valence compound. Solid state analogues are likely to be (A) insulators, (B) superconductors, (C) ‘normal’ metals. Case (D) illustrates the postulated explanation for the inverse isotope effect observed experimentally for palladium hydride (PdH, PdD, PdT). | ||
When the ZPE is too small, the system is localized (insulating, Fig. 4A). When the ZPE is too large, the system is delocalized (‘simple’ metal) and two distinct oxidation states cannot be recognized in the molecular (crystal) structure (Fig. 4C). We suggest that when the ZPE is close to EB, strong electron–phonon coupling dramatically influences the shape of the wavefunction and superconductivity may arise (Fig. 4B).
For PdH, electron transfer between Pd centers is greatly affected by motions of the hydride ligand.32 Hydrogen is the lightest ligand available to chemistry; here the ZPE can reach as much as 0.25 eV (for molecular H2). In cases when the ZPE for the proton isotope is larger than EB, and since the (classical) amplitude of vibrations—and also the vibrational zero point energy—decreases as the atomic mass increases for three isotopes of H, inverse isotope effect might indeed occur (Fig. 4D).
The case of PdH (small TC because of large energy of hydrogen motions (!), and concomitant easy mixing of d orbitals on Pd centers linked by H) brings to mind an idea for increasing the EB value by (i) involving in the electronic transport such orbitals, which are deeper lying (in energy) and more contracted than the 4d set of Pd (in this contribution we want to use 4f electrons), or by (ii) increasing the separation of the metal centers.35 It might allow a reversal of the atypical isotope effect, and also larger TC values by hydride superconductors. CsTmH3 and Cs2TmH4, which have a nearly filled yet significantly contracted f-shell, seem promising in terms of possible heavy-fermion superconductivity via appropriate chemical doping to these compounds; if EB proves to be too large, external pressure might be applied to bring these systems back towards the metal–insulator transition.
Cs2TmH4 (f13 TmII) is a quite formal analogue of other important systems with a nearly filled electron shell, namely Ln2CuO4 and Nd2CuO4 (d9 CuII). The latter two are parent (undoped) compounds of, respectively, hole-doped (La2−xBaxCuO4) and electron-doped (Nd2−xCexCuO4) oxocuprate superconductors. Simultaneously, CsTmH3 perovskite (f13 TmII) may be treated as a distant sibling of BaBiO3, with a distorted perovskite structure (s1 BiIV). The latter becomes Ba1−xKxBiO3, a regular perovskite superconductor, upon hole-doping.36
Thus, in terms of a formal electron count on the transition metal, Cs2TmII1−xYbIIxH4 would be equivalent to NdIII2−xCeIVxCuII/IO4, while Cs1−x□xTmII/IIIH3 (where □ stands for a vacancy)37 would be analogous to Ba1−xKxBiIV/VO3.38
We expect that tuning the transition between the f13 and f14 electron count39 within the LnH2 sublattice may bring Cs2TmII1−xYbIIxH4 and CsTmII1−xYbIIxH3 towards the anticipated metal/insulator borderline. At this stage, it is impossible to evaluate correctly from first principles whether these quaternary lanthanide hydrides will exhibit genuine heavy-fermion superconductivity, or plain ‘valence fluctuations’ (commonly observed for compounds of Yb, Tm, Eu, Sm, and also Ce and Tb).40,41 The consequences for the electronic and magnetic properties of novel hydride phases of the interplay of strong magnetism of 4f electrons and of the varying electron count (at the edge of the f shell closure), should be probed experimentally.
The estimated Debye temperature of CsYbH3 is substantial, ∼1800 K, while DOS below the Fermi level rises very steep, due to narrowness of the 4f electron band. The BCS theory predicts that these features alone are likely to prompt low-TC superconductivity in as yet unknown hole-doped hydrides of YbII, despite rather small involvement of H states into the DOS at the Fermi level of these compounds. Ultra-high pressure experiments constitute another prospective path of research. External pressure is an independent experimental variable which might enforce larger mixing of hydride 1s states into the quasi-valence 4f electrons and thus provoke larger involvement of the former in the electronic transport in these compounds. In this way a moderate-TC superconductivity could be achieved while making use of strong electron–phonon coupling of the states at the Fermi level with the high-frequency H-based phonons.
Conclusions
We predict the existence and some properties of four novel stoichiometric ternary hydrides of Tm and Yb. Our calculations indicate that—under easily achievable pressure of ∼20 GPa—CsLnH3, Ln = Yb, Tm, could be stable in the perovskite structure, while Cs2LnH4 might adopt the K2NiF4 one. These two structures are also shared by many 3D and 2D superconductors, and by some metallic hydrides.42 The electronic structure of these novel hydrides reveals a very narrow f band, very close to—or even interpenetrated by—the broad (s, p, d) conduction band. In addition, the phonon spectrum benefits from the high-frequency (up to 156 meV, 1800 K) modes involving motion of H atoms.43 The crystal structures adopted by hydrides in question, the isostructurality of YbII and TmII, and the electron count in the vicinity of shell closure (f13–f14) create the potential for tuning of electronic and magnetic properties of novel phases in the vicinity of the metal-to-insulator transition. An alluring possibility of valence fluctuations, or even heavy fermion superconductivity, arises.Acknowledgements
W.G. and T.J. want to thank the Interdisciplinary Center for Mathematical and Computational Modelling (ICM) at Warsaw University for providing access to its computational resources and financial support. The work at Cornell was supported by The Cornell Center for Materials Research and Cornell Summer Graduate Program. T.J. is grateful to the Ministry of Education and Sport for the Fellowship.References
- X. Chen, S. Lim, C. E. Plečnik, S. Liu, B. Du, E. A. Meyers and S. G. Shore, Inorg. Chem., 2004, 43, 692–698 CrossRef CAS.
- http://www.webelements.com; see also: R. Andreu and D. Pletcher, Electrochim. Acta, 2003, 48, 1065–1071 Search PubMed.
- G. Auffermann, Z. Anorg. Allg. Chem., 2002, 628, 1615–1618 CrossRef CAS.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283–1315 CrossRef CAS.
- See S. Harder, Angew. Chem., Int. Ed., 2004, 43, 2714–2718 Search PubMed and references therein.
- F. Gingl, K. Yvon and P. Fischer, J. Alloys Compd., 1993, 201, 105–107 CrossRef CAS.
- B. Huang, K. Yvon and P. Fischer, J. Alloys Compd., 1995, 227, 116–120 CrossRef CAS.
- B. Bertheville, H. Kohlmann, D. Sheptyakov and K. Yvon, J. Alloys Compd., 2003, 356, 128–132 CrossRef.
- Also YbIII (R = 1.008 Å) and TmIII (R = 1.020 Å) are nearly of the same size.
- Sandwich binuclear complexes of TmII are able to reduce N2 to N22−: W. J. Evans, N. T. Allen and J. W. Ziller, J. Am. Chem. Soc., 2001, 123, 7927–7928 Search PubMed.
- F. Gingl, T. Vogt, E. Akiba and K. Yvon, J. Alloys Compd., 1999, 282, 125–129 CrossRef CAS; W. Bronger and L. Breil, Z. Anorg. Allg. Chem., 1997, 623, 119–121 CrossRef CAS; B. Bertheville, T. Herrmannsdoerfer and K. Yvon, J. Alloys Compd., 2001, 325, 13–16.
- G. Q. Wu and R. Hoppe, Z. Anorg. Allg. Chem., 1983, 504, 55–59 CrossRef CAS; G. Q. Wu and R. Hoppe, Z. Anorg. Allg. Chem., 1984, 514, 99–106 CrossRef CAS.
- S. H. Wang, S. M. Luo and H. A. Eick, J. Less-Common Met., 1989, 149, 55–61 CrossRef.
- See ref. 194 in ref. 4.
- M. D. Segall, P. L. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169–11196 CrossRef CAS; G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS; G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- K. Parliński, Software Phonon, Cracow (2001), as implemented in MedeA 1.8, by Materials Design (2003); K. Parliński, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063–4066 Search PubMed.
- G. Meyer, Naturwissenschaften, 1978, 65, 258–258 CrossRef CAS.
- G. Schilling, C. Kunert, T. Schleid and G. Meyer, Z. Anorg. Allg. Chem., 1992, 618, 7–12 CAS.
- T. Schleid and G. Meyer, Z. Kristallogr., 1994, 209, 826–826 CAS.
- We use interchangeably the expressions ‘reaction energy’ and ‘reaction enthalpy’; earlier calculations for various hydride materials show that reaction enthalpy typically differs only very little from the ZPVE-corrected reaction energy, due to cancelling of these differences for all substrates and products together: E. Wimmer, Mater. Sci.—Poland, 2005, 23, 325–345 Search PubMed.
- E. Zintl and A. Harder, Z. Phys. Chem., Abt. B, 1931, 14, 265–284; C. E. Messer and P. C. Gianoukos, J. Less-Common Met., 1968, 15, 377–383 CrossRef CAS; J. E. Bonnet and J. N. Daou, J. Appl. Phys., 1977, 48, 964–968 CrossRef CAS; J. E. Bonnet and J. N. Daou, J. Phys. Chem., 1962, 66, 148–151 CrossRef.
- The reaction path might be different for hydrides of Yb and for those of Tm. For Tm compounds, the pressure increase should lead first to CsTmH3 (at 16.7 GPa) and then subsequently to Cs2TmH4 (at 19.7 GPa). For Yb compounds, however, CsYbH3 will be difficult to reach, as formation of (Cs2YbH4 + YbH2) is preferred over the whole pressure window studied. CsYbH3 might possibly be obtained via controlled thermal decomposition of CsYbIIIH4.
- N. W. Ashcroft and N. D. Mermin, Solid State Physics, Holt, Rinehart and Winston, New York, 1976, p. 308–309 Search PubMed.
- Modelling valence transitions in lanthanides from first principles is very difficult and resource-consuming. See for example: A. Delin, L. Fast, B. Johansson, J. M. Wills and O. Eriksson, Phys. Rev. Lett., 1997, 79, 4637–4640 Search PubMed; W. M. Temmerman, Z. Szotek, A. Svane, P. Strange, H. Winter, A. Delin, B. Johansson, O. Eriksson, L. Fast and J. M. Wills, Phys. Rev. Lett., 1999, 83, 3900–3903 CrossRef CAS.
- W. Grochala and R. Hoffmann, Angew. Chem., Int. Ed., 2001, 40, 2743–2781 CrossRef CAS; W. Grochala, J. Mol. Model., 2005, 11, 323–329 Search PubMed; J. Feng, W. Grochala, T. Jaroń, R. Hoffmann, A. Bergara and N. W. Ashcroft, Phys. Rev. Lett. Search PubMed , in press; M. Martinez-Canales, A. Bergara, J. Feng and W. Grochala, J. Phys. Chem. Solids Search PubMed , in press.
- The cut-off energy of the phonon spectrum (156 meV) is, of course, much smaller than that of the main vibron of solid H2 (around 546 meV). This comes from the reduced mass factor (beneficial by ∼1.4 times for H2) and from the smaller force constant for stretching of ionic Yb–H bonds than of strong H–H ones (by a factor of ∼2.5).
- We have not calculated the phonon dispersion and phonon DOS for the other ternary hydrides in question (CsTmH3, Cs2YbH4, Cs2TmH4). One can anticipate that these compounds will exhibit only small changes of phonon DOS as compared to CsYbH3. For example, Cs2YbH4 has two sets of Yb–H bonds: four longer ones at 2.305 Å and two shorter ones at 2.301 Å, while CsYbH3 has six equivalent Yb–H bonds at 2.305 Å. It means, that the Yb–H stretching mode (triply degenerate at Γ for cubic CsYbH3) will now split into a doubly degenerate in-plane mode (of similar frequency as for CsYbH3), and a non-degenerate mode of slightly higher frequency (connected with the Yb–H stretching in the crystallographic c direction). The former modes are responsible for the intra-layer electron–phonon coupling, and the latter mode for the inter-layer coupling. Additional minor splits are expected due to doubling of the Cs2YbH4 chemical formula in the unit cell of this compound (Z = 2).
- R. S. Liu, P. P. Edwards, S. F. Hu, D. A. Jefferson, Y. T. Huang, S. F. Wu and M. J. Tsai, J. Electron. Mater., 1993, 22, 1199–1203 Search PubMed; D. Meddeb, S. Charfi-Kaddour, M. Heritier and R. Bennaceur, Physica C, 2004, 408–410, 152–153 CrossRef CAS; J. Halbritter, J. Supercond., 2003, 16, 833–841 CrossRef CAS; M. S. Osofsky, R. J. Soulen, J. H. Claassen, G. Trotter, H. Kim and J. Horwitz, Phys. Rev. B, 2002, 66 Search PubMed Art. 020502 J. H. Jung, S. A. Seo, T. W. Noh, P. A. Sharma, N. Hur and S. W. Cheong, Solid State Commun., 2003, 126, 175–179 Search PubMed.
- J. Bardeen, L. N. Cooper and J. R. Schrieffer, Phys. Rev., 1957, 108, 1175–1204 CrossRef CAS.
- T. Skośkiewicz, Phys. Status Solidi A, 1972, 11, K123 CrossRef CAS ; PdHx shows inverse isotope effect: TC = 8 K for protide, 10 K for deuteride, and 12 K for tritide: B. Stritzker, Z. Phys., 1972, 257, 1–8 Search PubMed; J. E. Schirber, J. M. Mintz and W. Wall, Solid State Commun., 1984, 52, 837–838 CAS; H. Hemmes, A. Driessen, R. Griessen and M. Gupta, Phys. Rev. B, 1989, 39, 4110–4118 CrossRef CAS; T. Skośkiewicz, A. W. Szafrański, W. Bujnowski and B. Baranowski, J. Phys. C, 1974, 7, 2670–2676 CrossRef CAS . It is argued that the influence of zero-point motions and anharmonicity accounts for this unusual behavior: M. Yussouff, B. K. Rao and P. Jena, Solid State Commun., 1995, 94, 549–553 CrossRef CAS; B. M. Klein and R. E. Cohen, Phys. Rev. B, 1992, 45, 12405–12414 Search PubMed.
- Nonstoichiometric Pd hydride is very difficult to desribe using classical terms of chemistry such as cation and anion, or metal and ligand. In fact, some descriptions prefer the ‘solvated proton’ scenario, i.e., H+ is solvated by Pdδ−. In chemical terms, the first ionization potential of Pd (804 kJ mol−1) is smaller than that of H (1312 kJ mol−1), while the first electron affinity of Pd (53.7 kJ mol−1) is also smaller than that of H (72.8 kJ mol−1). Both properties point rather to a Pdδ+/Hδ− polarization in a palladium hydride, but the Pd–H bonding should of course be quite covalent.
- M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CAS.
- See ref. 26 and compare with: S. Larsson and A. Klimkãns, Int. J. Quantum Chem., 2000, 80, 713–720 Search PubMed.
- Note, conductivity and superconductivity usually requires presence of atomic bridges between the electron-exchanging centers in the crystal structure of an extended solid. Here we postulate that, paradoxically, certain molecular hydride materials might be even better superconductors than those hydrides which contain the direct M–H–M bridges.
- The hydride ligand is unique among all inorganic ligands, as it has no p orbitals and therefore π bonding is not available. This is important in terms of hole-doping, and progressive depopulation of the 1s2 band, as compared to analogous situations in oxides. Introducing holes into the hydride anion band by necessity means substantial weakening and even rupture of the element–hydrogen bond, in contrast to anions carrying p orbitals. This feature of H− also helps to explain the low-TC value of Th4H15. Th4H15 is formally a nonstoichiometric e−-doped (ThH4)4 (f0); an extra f1 electron thus needs to sit in a band originating from Th–H nonbonding states, built predominantly from Th 5f orbitals. Thus, conducting electron density is delocalized predominantly on Th network, with no contribution of H; the metallic form of Th4H15 (a ‘low-D metal’ in Zaanen–Sawatzky–Allen diagram) cannot fully benefit from high-frequency H-based phonons. In contrast, the f13/f14 doping for our systems introduces changes in a (weakly antibonding) σ* Ln–H states; the vibronic effects should thus be larger than for the f0/f1 perturbation.
- Here, two important differences between hole-doping to CuII and to TmII come to mind. Hole-doping (which proved very successful in introducing superconductivity in oxocuprates), might be realized for TmII compounds by their partial oxidation to trivalent Tm. Note, however, that, first, f12 TmIII is a strongly magnetic ion for most ligand fields (magnetic moment of ∼7.5 µB), in contrast to classical low-spin d8 CuIII for oxocuprate superconductors, and, second, the magnetic moment of f13 TmII itself (∼4.5 µB) is much greater than that of d9 CuII (∼1.8 µB). This means that magnetic ordering will be much easier for doped compounds of Tm than for those of Cu. Instead of introducing vacancies, other variations can also be used, e.g., LnIIH2 layer could be sandwiched between BaIIO sheets, and then doped by substitution at BaII site with either KI (hole-doping) or with AcIII (electron-doping).
- The electron doped BaPb1−xBixO3 superconductor does not have an isostructural lanthanide analogue. Hypothetical □1YbIII1−xTmIIIxH3 might be formally treated as such; unfortunately, parent stoichiometric YbH3 has not been synthesized. Also La2−xBaxCuO4 does not have any counterpart among the hydrides of Tm and Yb.
- Mixed EuII/YbII and SmII/YbII but not TmII/YbII compounds have been obtained; see ref. 12.
- D. T. Adroja and S. K. Malik, J. Magn. Magn. Mater., 1991, 100, 126–138 CrossRef CAS; J. F. Herbst and J. W. Wilkins, Phys. Rev. B, 1982, 26, 1689–1701 CrossRef CAS; B. Batlogg, A. Schlegel and P. Wachter, Physica B+C, 1977, 86, 229–230 CrossRef.
- Valence fluctuations have been linked to superconductivity in the early days of high-TC superconductivity; see for example: W. Y. Liang, J. Phys. C, 1987, 20, L571–L576 Search PubMed and have also inspired its very discovery.
- T. Sato, D. Noréus, H. Takeshita and U. Häussermann, J. Solid State Chem., 2005, 178, 3385–3392.
- Theoretical investigations of hypothetical LnBeH4, containing light Be, and of ternary hydrides of trivalent lanthanides, are now in progress in our laboratory.
Footnote |
| † Electronic supplementary information (ESI) available: Full DOS for CsYbH3 and VASP results. See DOI: 10.1039/b514773e |
| This journal is © The Royal Society of Chemistry 2006 |