DOI:
10.1039/B514359D
(Paper)
J. Mater. Chem., 2006,
16, 1431-1438
Highly efficient green light emitting polyfluorene incorporated with 4-diphenylamino-1,8-naphthalimide as green dopant
Received
10th October 2005
, Accepted 5th January 2006
First published on 25th January 2006
Abstract
The dopant/host methodology, which enables efficient tuning of emission color and enhancement of the electroluminescence (EL) efficiency of organic light emitting diodes (OLEDs) based on small molecules, is applied to the design and synthesis of highly efficient green light emitting polymers. Highly efficient green light emitting polymers were obtained by covalently attaching just 0.3–1.0 mol% of a green dopant, 4-(N,N-diphenyl) amino-1,8-naphthalimide (DPAN), to the pendant chain of polyfluorene (the host). The polymers emit green light and exhibit a high photoluminescence (PL) quantum yield of up to 0.96 in solid films, which is attributed to the energy transfer from the polyfluorene host to the DPAN dopant unit. Single layer devices (device configuration: ITO/PEDOT/Polymer/Ca/Al) of the polymers exhibit a turn on voltage of 4.8 V, luminance efficiency of 7.43 cd A−1, power efficiency of 2.96 lm W−1 and CIE coordinates at (0.26, 0.58). The good device performance can be attributed to the energy transfer and charge trapping from the polyfluorene host to the DPAN dopant unit as well as the molecular dispersion of the dopant in the host. The device performance is fairly comparable to that of state-of-the-art green light emitting poly(fluorene-co-benzothiadiazole), indicating that the covalently attached dopant/host polymer system is very promising for the development of highly efficient electroluminescent polymers of tunable emission color.
Introduction
Electroluminescent polymers have received much attention in academia and industry due to their applications in the generation of new display technologies.1–4 Among them, fluorene-based polymers have attracted particular attention in recent years because of their high photoluminescence (PL) and electroluminescence (EL) efficiency, good thermal and chemical stability, and color tunability within the entire visible spectrum.1–4 Normally, polyfluorene emits blue light with large band gap. In order to tune the emission color of polyfluorene, a common and successful approach is to incorporate narrow bandgap units into the main chain of polyfluorene.5–19 The narrow bandgap units are always aromatic heterocycles, such as benzothiadiazole,5–8 naphthoselenadiazole,14 bithiophene,18etc. In particular, poly(fluorene-co-benzothiadiazole) emits green light with good electron injection and transporting properties. Devices of poly(fluorene-co-benzothiadiazole) exhibit high luminance efficiency and long lifetime.5–8
The dopant/host emitter system is a common strategy to tune the emission color of organic light emitting diodes (OLEDs) based on small molecules.20–23 The energy transfer and charge trapping from the host to the dopant molecule allows the emission to originate from the dopant and the emission properties are dominated by the dopant. Moreover, the dopant/host system is found to enhance the EL efficiency and stability of OLEDs.22 This strategy has also been applied to tune the emission color of electroluminescent polymers.24–26 By covalently attaching the dopant unit (perylene derivatives) to a polymer host (polyfluorene) to form a single polymer, Ego et al have elegantly tuned the emission color of polyfluorene from blue to green, orange and red.24 The great success of dopant/host OLEDs based on small molecules22 also directed our research towards electroluminescence polymers with the dopant/host system. Recently, we have developed colorfast and efficient blue electroluminescent polymers with this system.27 Also, we have adopted the dopant/host system to achieve white emission from a single polymer.28–30 In this article, we report a novel highly efficient green light emitting polyfluorene with the dopant/host system.
4-(N, N-Diphenyl)amino-1,8-naphthalimide (DPAN), a derivative of the dye 1,8-naphthalimide,31,32 was selected as the dopant because of its high PL quantum yield. Moreover, DPAN has the ability to form a dopant/host system with polyfluorene. The absorption spectrum of DPAN sufficiently overlaps with the emission spectrum of polyfluorene, favouring the Förster energy transfer from polyfluorene to DPAN.33 Also, the LUMO and HOMO energy levels of DPAN lie between those of polyfluorene, favouring the charge trapping of the DPAN dopant unit.34,35 The dopant molecule was attached to the pendant chain of polyfluorene by an alkyl spacer with a low content of 0.3–1 mol%, avoiding the quenching effect of the dopant unit. This molecular design brings the molecular dispersion of the dopant in the host,24 which is different from blending36–38 the dopant with the host because the former case can achieve higher efficiency and better spectral stability without phase separation.27–30 A photoluminescence quantum yield up to 0.96 was observed for the neat films of the polymers. To our best knowledge, these materials are the most efficient light-emitting polymers reported to date. Polymer light emitting diodes (PLEDs) with a non-optimized single-layer device structure of ITO/PEDOT/polymer/Ca/Al have been fabricated and a high luminance efficiency of 7.43 cd A−1 with CIE coordinates of (0.26, 0.58) is demonstrated. This performance is fairly comparable to that of the state-of-the-art green light emitting poly(fluorene-co-benzothiadiazole) in similar device configurations,5 and indicates that polyfluorenes of the dopant/host system are very promising for the development of highly efficient electroluminescent polymers with tunable emission color.
Result and discussion
Synthesis and characterization
As shown in Scheme 1, the 4-diphenylylamino-1,8-naphthalimide dopant unit was first attached to a phenyl ring and then copolymerized into polyfluorene by Suzuki condensation39 with different feed ratios of the three monomers. In the copolymers, the DPAN was attached to the pendant chain of polyfluorene without affecting the electronic properties of polyfluorene backbone. All the polymers were thoroughly purified before characterization and device fabrication. Structural characterization of the polymers was carried out with 1H NMR, 13C NMR and elemental analysis. In the 1H NMR spectra, the signals at 4.60, 4.32 and 3.97 ppm are all attributed to the DPAN unit, from which we can calculate its actual content. For PFG40, the exact content of DPAN unit is 0.033, which is close to the feed ratio of 0.040 of the monomer 5, indicating that the incorporation of DPAN unit into polyfluorene is very efficient in the polymerization process. For PFG3, PFG5, PFG10, the contents of DPAN unit are too low to give accurate contents from 1H NMR spectra due to experimental error. Therefore, the actual content of DPAN in the polymers is assumed to be the same with the feed ratios. For comparison, we have also synthesized poly(9,9-dioctylfluorene-2,7-yl) homopolymer (PF) and the model compound 4-(N, N-diphenyl)amino-9-decyl-1,8-naphthalimide (GMC). The number-average molecular weight (Mn) of the polymers, determined by gel permeation chromotagraphy (GPC) with polystyrene as standard, is in the range of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 to 28000 with the polydispersity from 1.56 to 1.87 (Table 1). All these polymers show good thermal stability with thermal decomposition temperature higher than 430 °C. All these polymers are soluble in common organic solvent, such as chloroform, toluene and THF.
200 to 28000 with the polydispersity from 1.56 to 1.87 (Table 1). All these polymers show good thermal stability with thermal decomposition temperature higher than 430 °C. All these polymers are soluble in common organic solvent, such as chloroform, toluene and THF.
 |
| | Scheme 1 Synthetic route for monomers and polymers. Reagents and conditions: (i) 1-bromooctane, KOH, ethanol, reflux; (ii) 1,2-dibromoethane, KOH (aq., 2 M), tetra(n-butyl)ammonium bromide, toluene, 90 °C; (iii) bromine, dichloromethane; (iv) KOH, DMSO, 120 °C; (v) iodobenzene, K2CO3, CuI, 18-crown-6, DMPU, 190 °C; (vi) Pd(PPh3)4, Aliquat 336, toluene, K2CO3 (aq., 2 M), 90 °C; (vii) 1-bromodecane, KOH, DMSO, 120 °C. | |
Table 1 The content of DPAN unit, number average molecular weight (Mn), polydispersity (PDI), thermal decomposition temperature (Td), photoluminescence quantum efficiency (ΦPL) of PF, PFG3, PFG5, PFG10 and PFG40
| Polymer |
Content |
Mn |
PDI |
Td |
Φ
PL
|
|
PF
|
0
|
28000 |
1.87 |
434 |
0.45 |
|
PFG3
|
0.003
|
25600 |
1.65 |
432 |
0.89 |
|
PFG5
|
0.005
|
24800 |
1.69 |
432 |
0.96 |
|
PFG10
|
0.01
|
20200 |
1.56 |
433 |
0.83 |
|
PFG40
|
0.04
|
18600 |
1.86 |
431 |
0.23 |
Electrochemical properties
Cyclic voltammetry of all polymers in solid films was carried out in acetonitrile with n-Bu4NClO4 (0.10 M) as the electrolyte. PFG3, PFG5, PFG10 all exhibited identical redox behavior with that of PF, indicating that the incorporation of small amount of DPAN has negligible effect on the redox properties of the resulting polymers. Fig. 1 shows the cyclic voltagramms of PF and the model compound GMC. Based on the onset potential of oxidation and reduction process, and according to the formula (EHOMO = −(Eox
+ 4.34) eV and ELUMO = −(Ered
+ 4.34) eV),40 we can estimate the HOMO and LUMO energy levels of PF and GMC. The HOMO and the LUMO levels of GMC are −5.61 and −3.20 eV, respectively, and are above and below the corresponding values of PF that exhibit a HOMO level of −5.81 eV and a LUMO level of −2.12 eV, indicating possible charge trapping of the DPAN units in the electroluminescence process.34,35
 |
| | Fig. 1 Cyclic voltagramms of PF and GMC. | |
Photophysical properties
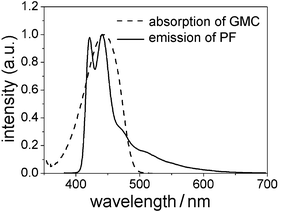
For photophysical measurements, a polymer film with a thickness of approximately 70 nm was spin-coated on quartz from a chloroform solution (10 mg ml−1). Similar to the results obtained by cyclic voltammetry characterization, all the DPAN-containing polymers have identical UV-Vis absorption spectra with PF with an absorption maximum at 393 nm attributed to the π–π* transition of the polymer backbone (Fig. 2). No absorption by the DPAN unit (λmax = 444 nm for the model compound as shown in Fig. 3) is observed. In contrast, the PL spectra of the copolymers with an excitation at 380 nm, as illustrated in Fig. 4a, exhibit a strong emission peak from DPAN at 520 nm and only a weak emission peak from the polyfluorene backbone at 420/440 nm. The weak emission peaks from polyfluorene backbone decrease with increasing content of naphthalimide units from 0.3 to 1.0 mol%. The incorporation of DPAN into polyfluorene leads to little change in the absorption spectra but results in a great change in the PL spectra. This is due to the Förster energy transfer from the polyfluorene backbone to the DPAN unit because of the efficient overlap of the absorption spectrum of GMC with PL spectrum of PF (see Fig. 3).33 The PL spectrum of PFG40 is dominated by total emission from the DPAN unit with the peak at 532 nm. This redshift results from the interaction of the DPAN unit due to its high content.
 |
| | Fig. 4 Photoluminescence (a) and electroluminescence (b) spectra of polymers with different contents of DPAN unit. | |
Photoluminescence quantum yield (ΦPL) of the polymers and GMC in the solid states were measured by means of an integrated sphere (according to the method of ref. 41) with an excitation at 409 nm (Table 1). GMC dispersed in PMMA film with a concentration of 10−3 M gave a quantum yield of 0.90, which is comparable to that of other efficient light-emitters. As listed in Table 1, the ΦPL value of the four copolymers, PFG3, PFG5,PFG10 and PFG40, in neat films is 0.89, 0.96, 0.83 and 0.23, respectively. The low photoluminescence quantum yield of PFG40 is due to the quenching effect of the dopant unit because of its high content. The ΦPL value of PFG3, PFG5,PFG10 is much higher than that of PF, 0.45, and other fluorene-based polymers ever reported.5–24 These remarkable ΦPL of PFG3, PFG5,PFG10 is attributed to the Förster energy transfer from polyfluorene to the DPAN unit and the high PL efficiency of the DPAN unit. This result also indicates that a low content of dopant unit is important to achieve high PL efficiency of the resulting polymers. The relatively lower ΦPL value at 0.3 and 1.0 mol% is possibly due to the incomplete energy transfer in the former case and the presence of concentration quenching in the latter case.
Electroluminescence properties
To investigate the EL properties of the polymers, single-layer devices were fabricated with a configuration of ITO/PEDOT(50 nm)/polymer(70 nm)/Ca(10 nm)/Al(100 nm). PFG40 was not further characterized because of its low PL quantum yield. The polymer layer was prepared following the same procedure for preparation of polymer films for photophysical measurements; except that PEDOT treated ITO glass substrates were used. The EL spectra of the polymers are illustrated in Fig. 4b. PFG3, PFG5,PFG10 all exhibit predominant green emission from the DPAN units and only negligible blue emission from the polyfluorene backbone, indicating that only a very small amount of dopant unit is needed to tune the emission color of polyfluorene. Comparison of the EL and PL spectra of the same polymer indicates that the contribution of blue emission from the polyfluorene backbone in the EL spectrum is much smaller compared to the PL spectrum. This difference is attributed to different mechanisms in the EL and PL processes. In the former case, both charge trapping and energy transfer contribute to the emission from DPAN. However, in the latter case, charge trapping does not occur and only energy transfer is involved. Color purity of these copolymers was excellent, indicated by their CIE coordinates (see Table 2) and a narrow full width at half maximum (FWHM) of 66 nm. For example, the CIE coordinates (0.26, 0.58) of PFG3 are very close to those of the standard green (0.26, 0.65) demanded by the National Television System Committee (NTSC).
Table 2 Electroluminescence performance of the devices of PF, PFG3, PFG5, and PFG10
| Polymer |
Onset voltage/V |
Maximum brightness/cd m−2 |
Luminous efficiency/cd A−1 |
Power efficiency/lm W−1 |
CIE (x,y) |
|
PF
|
3.2 |
2201 |
0.84 |
0.58 |
(0.16, 0.09) |
|
PFG3
|
4.8 |
21595 |
7.43 |
2.96 |
(0.26, 0.58) |
|
PFG5
|
5.4 |
17596 |
6.18 |
2.30 |
(0.27, 0.59) |
|
PFG10
|
8.2 |
15639 |
4.48 |
1.27 |
(0.28, 0.60) |
The performance data of all the devices are listed in Table 2. The device of the homopolymer PF exhibited a turn-on voltage of 3.2 V, whereas the copolymers exhibited turn on voltages of 4.8 to 8.2 V with increasing DPAN content from PFG3 to PFG10. The increase of the turn-on voltage with increasing DPAN concentration is due to the charge trapping of the DPAN unit. At low operating voltages, the electrons (holes) injected from the cathode (anode) are trapped by the DPAN unit in the region near the cathode (anode) and cannot migrate to recombination with each other. Therefore, higher voltages are needed for the device to emit light. A luminance efficiency of 0.84 cd A−1, a power efficiency of 0.58 lm W−1 and a maximum brightness of 2201 cd m−2 for PF are much higher than those of polyfluorene homopolymer reported by other groups42 and are comparable with those of triaryl amine end-capped polyfluorenes in similar device structures.43 All devices based on the copolymers exhibited efficient green emission with luminance efficiency ranging from 4.48 to 7.43 cd A−1. A weak dependence of luminance efficiency on current density was observed (see Fig. 5). In particular, the device fabricated with PFG3 has a turn on voltage of 4.8 V, a current efficiency of 7.43 cd A−1, a power efficiency of 2.96 lm W−1 and a maximum brightness of 21595 cd m−2 (see Fig. 6). These values are comparable to those of poly(fluorene-co-benzothiadiazole).5 It is worth noting that the content of DPAN in PFG3 is only 0.3 mol% and that an increase of DPAN's content leads to an increase of the onset voltage and a decrease of EL efficiency. Hence, our result indicates that a low content of dopant unit is enough to tune the emission color of polyfluorene and is crucial to realize good EL performance of the devices.
 |
| | Fig. 5 Dependence of luminance efficiency on current density of the devices based on polymers with different contents of DPNA unit. | |
 |
| | Fig. 6 Voltage–current density–brightness curve of the device based on PFG3. | |
The excellent EL performance of PFG3 is principally attributed to the energy transfer and charge trapping from polyfluorene to highly fluorescent DPAN unit, which enhances the radiative decay rate of singlet excitons. In addition, the charge trapping of the DPAN units also enhances the EL efficiency by increasing the recombination probability of charge carriers. The electron and/or holes trapped by DPAN units causes a build up in local charge density, which enhances the possibility of attracting oppositive charge carriers. That is, DPAN offers an effective recombination site for electrons and holes.
In order to evaluate the stability of these polymers, the device of PFG3 was encapsulated with a cover glass and epoxy and the performance is shown in Fig. 7. At a constant current, the brightness decreases and the voltage is observed to increase from an initial brightness of 800 cd m−2. After this rapid reduction of brightness and increase of driving voltage at the beginning, the device performance almost levels off at approximately 50 min. Most importantly, during the operation, the EL spectra remained unchanged and the CIE coordinates kept within the range of (0.260–0.261, 0.581–0.582).
 |
| | Fig. 7 Dependence of the brightness and voltage on the operation time of the device of PFG3 under constant current stress. | |
In comparison, we fabricated another device with 0.3 mol% GMC blended with PF as emissive materials. The device showed the turn on voltage of 5.0 V and maximum luminance efficiency of 4.62 cd A−1 and power efficiency of 1.82 lm W−1. This performance is inferior to that of PFG3, indicating that the molecular dispersion of dopant in the host material is important to realize highly efficient PLEDs.
Conclusion
In summary, we have developed a series of highly efficient green light emitting polyfluorenes by covalently attaching very small amounts of a highly fluorescent dopant unit, 4-(N, N-diphenyl)amino-1,8-naphthalimide, to the pendant chain of polyfluorene. The attachment of 0.3–1 mol% of dopant unit results in little change of the absorption spectrum and electrochemical properties of the resulting polymers but results in a great change in the emission properties due to energy transfer and charge trapping from polyfluorene to DPAN unit. The resulting polymers exhibits high photoluminescence quantum yields in solid films. Non-optimized single layer device of these polymers achieved a luminance efficiency of 7.43 cd A−1, which is fairly comparable to those of the state-of-art green light-emitting poly(fluorene-co-benzothiadiazole) with similar device structure. Both Förster energy transfer and charge trapping and high photoluminescence quantum yields of the copolymers as well as molecular dispersion of the dopant are responsible for the efficient green light emission. Our results indicate that polyfluorene with dopant/host system is very promising for achieving excellent electroluminescence performance with tunable emission color.
Experimental
Materials
All the reagents and solvents used for the syntheses were purchased from Aldrich and Acros companies and used without further purification except for tetrahydrofuran (THF), which was dried over sodium/benzophenone. 2,7-dibromo-9,9′-dioctylfluorene (6), 2,7-bis(trimethylene boronate)-9,9′-dioctylfluorene (7) were synthesized according to the procedure from the literature.25 All reactions were performed under a dry argon atmosphere.
A mixture of hydroquinone (11.0 g, 100 mmol) and potassium hydroxide (11.2 g, 200 mmol) in 200 ml ethanol was heated under reflux with stirring. 1-Bromooctane (15.44 g, 80 mmol) in 20 ml ethanol was added dropwise to the stirred solution. The mixture had been refluxed for 14 h. After cooled to room temperature, the mixture was poured into 500 ml 1 M HCl and extracted with chloroform. The organic layer was washed with brine and dried over anhydrous sodium sulfate. After the solvent had been removed by rotary evaporation, the residual was recrystallized from hexane to afford the titled compound as a white crystal. Yield: 11.2 g (63%). 1H NMR (300 MHz, CDCl3) δ (ppm): 6.78 (m, 4H), 4.39 (br, 1H), 3.89 (t, 2H, J = 6.5 Hz), 1.75 (m, 2H), 1.50–1.20 (m, 10H), 0.88 (t, 3H, J = 6.9 Hz). Anal. calcd: C, 75.63; H, 9.97. Found: C, 74.27; H, 9.70.
A mixture of 1,2-dibromoethane (18.8 g, 100 mmol), 4-(octyloxy)phenol (1) (4.44 g, 20 mmol), tetra(n-butyl)ammonium bromide (0.32 g, 1 mmol), 50 ml toluene and 50 ml 2 M aqueous potassium hydroxide was stirred at 90 °C for 12 h. After cooled to room temperature, the mixture was extracted with chloroform. The organic layer was washed with water and dried over anhydrous sodium sulfate. After the solvent had been removed by rotary evaporation, the residual was recrystallized from ethanol to afford the titled compound as a white crystal. Yield: 3.16 g (48.4%). 1H NMR (300 MHz, CDCl3) δ (ppm): 6.84 (m, 4H), 4.23 (t, 2H, J = 6.3 Hz), 3.90 (t, 2H, J = 6.6 Hz), 3.61 (t, 2H, J = 6.3 Hz), 1.75 (m, 2H), 1.50–1.20 (m, 10H), 0.88 (t, 3H, J = 6.9 Hz). Anal. calcd: C, 58.36; H, 7.65. Found: C, 58.18; H, 7.23.
1,4-Bibromo-2-(octyloxy)-5-(2-bromoethyloxy)benzene (3)
A solution of 1-(2-bromoethyloxy)-4-(octyloxy)benzene (2) (3.29 g, 10 mmol) in 50 ml dichloromethane was stirred under room temperature. Bromine (4.80 g, 30 mmol) in 20 ml dichloromethane was added dropwise. The mixture was stirred under dark for 10 h. Then the mixture was washed subsequently with aqueous sodium hydrogen sulfate and brine, and then dried over anhydrous sodium sulfate. After the solvent had been removed by rotary evaporation, the residual was recrystallized from ethanol to give the titled compound as a white crystal. Yield: 4.11 g (84%). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.09 (s, 2H), 4.28 (t, 2H, J = 6.3 Hz), 3.95 (t, 2H, J = 6.6 Hz), 3.73 (t, 2H, J = 6.3 Hz), 1.75 (m, 2H), 1.50–1.20 (m, 10H), 0.91 (t, 3H, J = 6.9 Hz). 13C NMR (300 MHz, CDCl3) δ (ppm): 154.0, 152.1, 68.9, 68.6, 31.8, 29.4, 29.3, 26.1, 22.7, 14.11. Anal. calcd: C, 39.45; H, 4.76. Found: C, 40.00; H, 4.30.
1,4-Dibromo-2-(octyloxy)-5-(2-(4-amino-1,8-naphthalimide-9-yl)ethyloxy)benzene (4)
Into a stirred solution of 4-amino-1,8-naphthalimide (2.12 g, 10 mmol) in 60 ml DMSO was added potassium hydroxide powder (0.56 g, 10 mmol). After 10 min, 1,4-dibromo-2-(octyloxy)-5-(2-bromoethyloxy) benzene (3) (5.38 g, 11 mmol) was added and the mixture was stirred at 120 °C for another 3 h. After cooling to room temperature, the mixture was poured into water and extracted with ethyl acetate. The organic layer was separated and washed with brine and then dried over anhydrous sodium sulfate. After the solvent had been removed by rotary evaporation, the dark yellow solid was purified with column chromatography on silica gel to afford the titled compound as a yellow solid. Yield: 4.39 g (71%). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.63 (d, 1H, J = 7.3 Hz), 8.45 (d, 1H, J = 8.1 Hz), 8.12 (d, 1H, J = 8.4 Hz), 7.68 (t, 1H, J = 7.9 Hz), 7.20 (s, 1H), 7.01 (s, 1H), 6.90 (d, 1H, J = 8.1 Hz), 4.65 (t, 2H, J = 6.1 Hz), 4.29 (t, 2H, J = 6.1 Hz), 3.90 (t, 2H, J = 6.5 Hz), 1.79 (m, 2H), 1.70–1.20 (m, 10H), 0.88 (t, 3H, J = 7.0 Hz). 13C NMR (300 MHz, CDCl3) δ (ppm): 163.6, 162.9, 149.4, 148.5, 148.4, 132.9, 130.6, 128.8, 126.1, 123.9, 121.8, 119.0, 117.8, 117.3, 110.6, 110.3, 110.1, 108.5. 69.2, 66.0, 37.6, 30.7, 28.2, 28.1, 24.9, 21.6, 13.1. The signal at 37.6 ppm in 13C NMR indicates that the alkylation occurs at the imide nitrogen not the primary amine. Anal. calcd: C, 54.39; H, 4.89; N, 4.53. Found: C, 54.28; H, 4.71; N, 4.82. Electrospray ionization mass spectrum (ESI-MS): [M + H]+: calcd: 617.1; found: 617.4.
1,4-Dibromo-2-(octyloxy)-5-(2-(4-diphenylamino-1,8-naphthalimide-9-yl)ethyloxy)benzene (5)
A mixture of 1,4-dibromo-2-(octyloxy)-5-(2-(4-amino-1,8-naphthalimide-9-yl)ethyloxy)benzene (4) (0.618 g, 1.0 mmol), iodobenzene (1.020 g, 5.0 mmol), potassium carbonate (0.414 g, 3.0 mmol), 18-crown-6 (0.015 g, 0.05 mmol), copper iodide (0.038 g, 0.2 mmol), 0.30 mL DMPU was stirred at 190 °C for 48 h. After cooled to room temperature, the mixture was dissolved in chloroform and washed with brine. The organic layer was dried over anhydrous Na2SO4. After the solvent had been removed, the crude product was purified with column chromatography on silica gel to afford the titled compound as an orange-red solid. Yield: 0.151 g (19%). 1H NMR (300 MHz, CDCl3): δ (ppm) 8.55 (dd, 1H, J = 7.3 Hz, J = 0.8 Hz), 8.52 (d, 1H, J = 8.1 Hz) 8.18 (d, 1H, J = 8.5 Hz), 7.49 (dd, 1H, J = 8.5 Hz, J = 7.3 Hz), 7.37 (d, 1H, J = 8.0 Hz), 7.30–7.23 (m, 4H), 7.12–7.00 (m, 8H), 4.68 (t, 2H, J = 5.8 Hz), 4.30 (t, 2H, J = 5.8 Hz), 3.90 (t, 2, J = 6.1 Hz H), 1.77 (m, 2H), 1.50–1.25 (m, 10H), 0.88 (t, 3H, J = 7.0 Hz). 13C NMR (300 MHz, CDCl3) δ (ppm): 164.3, 163.7, 153.2, 152.4, 151.0, 148.4, 132.4, 131.5, 131.4, 130.4, 129.6, 129.4, 128.0, 126.3, 125.4, 124.2, 123.9, 123.2, 123.1, 122.8, 118.6, 118.3 , 70.3, 66.9, 38.9, 31.8, 29.7, 29.2, 29.1, 26.0, 22.7, 14.1. Anal. calcd: C, 62.35; H, 4.97; N, 3.64. Found: C, 62.12; H, 4.76; N, 3.53. ESI-MS: [M + H]+: calcd: 769.1; found: 769.4.
General procedure of Suzuki polymerization39
A mixture of 2,7-dibromo-9,9-dioctylfluorene (6), 2,7-bis(trimethylene boronate)-9,9′-dioctylfluorene (7), 1,4-dibromo-2-(octyloxy)-5-(2-(4-diphenylamino-1,8-naphthalimide-9-yl)ethyloxy)benzene (5) with corresponding feed ratio, Aliquat 336 (0.05 g, 0.12 mmol), tetrakis(triphenylphosphine)palladium (2.3 mg) under argon was added 2.5 ml 2 M aqueous potassium carbonate and 7 ml toluene. The mixture was stirred at 90 °C for 48 h and at 60 °C for another 24 h and then poured into methanol. The precipitate was collected by filtration, dried and then dissolved in dichloromethane. The solution was washed with water and dried over anhydrous Na2SO4. After most of the solvent had been removed, the residue was poured into stirred methanol to give a fiber-like solid. The polymer was further purified by extracting with acetone for 24 h. The reprecipitation procedure in dichloromethane–methanol was then repeated several times. The final product, a light yellow fiber, was obtained after drying in vacuum with a yield of 45–60%. PF1H NMR (300 MHz, CDCl3) δ (ppm) 7.87 (d, 2H), 7.72 (br, 4H), 2.10 (br, 4H), 1.14 (br, 24H), 0.81 (t, 6H). 13C NMR (300 MHz, CDCl3) δ (ppm): 151.4, 110.1, 139.7, 125.8, 121.1, 119.6, 55.0, 40.0, 31.4, 29.7, 28.8, 23.5, 22.2, 13.7. Anal. calcd: C, 89.69; H, 10.31. Found: C, 89.08; H, 10.02. PFG3, PFG5, PFG10 showed the similar 1H NMR and elemental analysis results with those of PF. PFG401H NMR (300 MHz, CDCl3) δ (ppm) 8.53 (br, 0.032H), 7.83 (d, 2H), 7.68 (br, 4H), 7.49 (br, 0.60H), 7.38 (br, 0.28H), 6.92 (br, 0.34H), 4.60 (br, 0.062H), 4.32 (br, 0.068H), 3.97 (br, 0.062H), 2.11 (br, 4H), 1.14 (br, 24H), 0.81 (t, 6H). Anal. calcd: C, 88.85; H, 10.00; N, 0.32. Found: C, 88.11; H, 9.71; N, 0.60.
4-Amino-9-N-decyl-1,8-naphthalimide (8)
Into a solution of 4-amino-1,8-naphthalimide (2.12 g, 10 mmol) in 60 ml DMSO was added potassium hydroxide (0.56 g, 10 mmol) at 120 °C. After 10 min, 1-bromodecane (2.21 g, 10 mmol) was added and the mixture was kept stirring for 12 h. After cooled to room temperature, the mixture was poured into water and extracted with ethyl acetate. The organic layer was collected and washed with brine and dried over anhydrous Na2SO4. The crude product was further purified by column chromatography on silica gel to give the title compound as a yellow solid. Yield: 2.50 g (71%). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.60 (d, 1H, J = 7.3 Hz), 8.42 (d, 1H, J = 8.4 Hz), 8.10 (d, 1H, J = 8.4 Hz), 7.66 (t, 1H, J = 7.8 Hz), 6.89 (d, 1H, J = 8.1 Hz),4.93 (br, 2H), 4.15 (t, 2H, J = 7.6 Hz), 1.74–1.18 (m,16H), 0.87 (t, 3H, J = 6.7 Hz). 13C NMR (300 MHz, CDCl3) δ (ppm): 163.5, 163.0 148.4, 132.7, 130.3, 128.7, 125.9, 123.8, 122.0, 119.0, 110.9, 108.4, 39.3, 30.8, 28.5, 28.4, 28.2, 27.2, 26.2, 21.6, 13.1. The signal at 39.3 ppm in 13C NMR indicates that the alkylation occurs at the imide nitrogen not at the primary amine. Anal. calcd: C, 74.97; H, 8.01; N, 7.95. Found: C, 74.43; H, 7.90; N, 8.20. Matrix-assisted laser desorption/ionization mass spectrometry (MALTI-TOF MS): calcd: 352.2; found: 352.2.
4-N,N-Diphenylamino-9-decyl-1,8-naphthalimide (GMC)
A mixture of 4-amino-9-decyl-1,8-naphthalimide (0.704 g, 2.0 mmol), iodobenzene (4.080 g, 20.0 mmol), potassium carbonate (0.828 g, 6.0 mmol), 18-crown-6 (0.030 g, 0.11 mmol), copper iodide (0.076 g, 0.4 mmol), 0.30 mL DMPU had been stirred at 190 °C for 12 h. After cooled to room temperature, the mixture was dissolved in chloroform. The organic solution was washed with brine and dried over anhydrous Na2SO4. The crude product was purified by column chromatography on silica gel to give the title compound as an orange-red solid. Yield: 0.796 g (79%). 1H NMR (300 MHz, CDCl3) δ (ppm): 8.55–8.48 (m, 2H), 8.17 (dd, 1H, J = 8.6 Hz, J = 1.0 Hz), 7.49 (dd, 1H, J = 7.3 Hz, J = 8.5 Hz), 7.37 (d, 1H, J = 8.0 Hz), 7.24 (m, 4H), 7.04 (m, 6H), 4.17 (t, 2H, J = 7.5 Hz), 1.18–1.74 (m,16H), 0.87 (t, 3H, J = 6.9 Hz). 13C NMR (300 MHz, CDCl3) δ (ppm): 163.2, 162.7, 149.7, 147.4, 131.0, 130.2, 130.0, 129.3, 128.5, 127.0, 125.3, 124.5, 122.8, 122.6, 122.3, 117.8, 39.4, 30.9, 28.5, 28.4, 28.3, 27.2, 26.2, 21.7, 13.1. Anal. calcd: C, 80.92; H, 7.19; N, 5.55. Found: C, 79.41; H, 7.15; N, 5.40. MALTI-TOF MS calcd: 504.2; found: 504.2.
Instruments
1H NMR and 13C NMR spectra were recorded with Bruke Avance 300 NMR spectrometer. The elemental analysis was performed using a Bio-Rad elemental analysis system. The number- and weight-average molecular weights of the polymers were determined by gel permeation chromotagraphy (GPC) with a Waters 410 instrument with polystyrene as standards and THF as eluent. UV-Vis absorption, photoluminescence and electroluminescence spectra were measured by Perkin-Elmer Lambda 35 Uv/Vis spectrometer and Perkin-Elmer LS50B spectrofluorometer, respectively. Cyclic voltammograms of polymer films on Pt plate were recorded on an EG&G 283 (Princeton Applied Research) at room temperature in a solution of n-Bu4NClO4 (0.10 M) in acetonitrile at a scan rate of 100 mV s−1. A Pt wire and an Ag/AgCl electrode were used as the counter electrode and the reference electrode, respectively. Thermal gravimetric analysis (TGA) was performed under a nitrogen atmosphere at a heating rate of 10 °C min−1 through the use of Perkin-Elmer-TGA 7. The current–voltage and brightness–voltage curves of devices were measured using a Keithley 2400/2000 current/voltage source unit calibrated with a silicon photodiode.
Device fabrication
The indium–tin oxide (ITO) glass plates were degreased in ultrasonic solvent bath and then dried in a heating chamber at the temperature of 120 °C. The poly(styrene sulfonic acid) doped poly(ethylenedioxythiophene) was spin-coated on the treated ITO at 3000 rms for 60 s and then baked for 15 min at 120 °C to give an approximate thickness of 50 nm. The polymer layer (approximately 70 nm) was then spin-coated onto the PEDOT/ITO coated glass substrate in chloroform solution (10 mg ml−1) at ambient atmosphere. Finally, a thin layer of calcium (10 nm) followed by a layer of aluminum (100 nm) is deposited in a vacuum thermal evaporator through a shadow mask at a pressure of 3 × 10−5 Torr. The active area of the diodes is 16 mm2.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 20574067) and 973 Project (2002CB613402). The authors thank Dr Aldred P. Matthew for fruitful discussion.
References
- A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402 CrossRef.
- M. T. Bernius, M. Inbasekaran, J. O'Brien and W. S. Wu, Adv. Mater., 2000, 12, 1737 CrossRef CAS.
- D. Neher, Macromol. Rapid Commun., 2001, 22, 1365 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS.
- M. Bernius, M. Inbasekaran, E. P. Woo, W. S. Wu and L. Wujkoski, J. Mater. Sci.: Mater. Electron., 2000, 11, 111 CrossRef CAS.
- H. Spreitzer, H. Becker, E. Breuning, A. Falcou, K. Treacher, A. Büsing, A. Parham, P. Stöβel, S. Heun and J. Steiger, Proc. SPIE – Int. Soc. Opt. Eng., 2003, 4800, 16 CAS.
- C. D. Müller, A. Falcou, N. Reckefuss, M. Rojahn, V. Wiederhirn, P. Rudati, H. Frohne, O. Nuyken, H. Becker and K. Meerholz, Nature, 2003, 421, 829 CrossRef.
- M. S. Liu, X. Jiang, S. Liu, P. Herguth and A. K.-Y. Jen, Macromolecules, 2002, 35, 3532 CrossRef CAS.
- S. Beaupré and M. Leclerc, Adv. Funct. Mater., 2002, 12, 192 CrossRef CAS.
- M. S. Liu, J. D. Luo and A. K.-Y. Jen, Chem. Mater., 2003, 15, 3496 CrossRef CAS.
- P. Sonar, J. Y. Zhang, A. C. Grimsdale, K. Müllen, M. Surin, R. Lazzaroni, P. Leclère, S. Tierney, M. Heeney and I. McCulloch, Macromolecules, 2004, 37, 709 CrossRef CAS.
- R. Q. Yang, R. Y. Tian, Q. Hou, W. Yang and Y. Cao, Macromolecules, 2003, 36, 7453 CrossRef CAS.
- Q. Hou, Y. S. Xu, W. Yang, M. Yuan, J. B. Peng and Y. Cao, J. Mater. Chem., 2002, 12, 2887 RSC.
- J. Yang, C. Y. Jiang, Y. Zhang, R. Q. Yang, W. Yang, Q. Hou and Y. Cao, Macromolecules, 2004, 37, 121.
- Q. Peng, Z. Y. Lu, Y. Huang, M. G. Xie, S. H. Han, J. B. Peng, Y. Cao and W. Huang, Macromolecules, 2004, 37, 260 CrossRef CAS.
- E. Lim, B.-J. Jung and H.-K. Shim, Macromolecules, 2003, 36, 4288 CrossRef CAS.
- N. S. Cho, D.-H. Hwang, B.-J. Jung, E. Lim, J. Lee and H.-K. Shim, Macromolecules, 2004, 37, 5265 CrossRef CAS.
- J. Pei, W.-L. Yu, W. Huang and A. J. Heeger, Chem. Commun., 2000, 1631 RSC.
- B. Liu, W.-L. Yu, Y.-H. Lai and W. Huang, Macromolecules, 2000, 33, 8945 CrossRef CAS.
- C. W. Tang, S. A. van Slyke and C. H. Chen, J. Appl. Phys., 1989, 65, 3610 CrossRef CAS.
- J. M. Shi and C. W. Tang, Appl. Phys. Lett., 1997, 70, 1665 CrossRef CAS.
- L. S. Hung and C. H. Chen, Mater. Sci. Eng., R, 2002, 39, 143 Search PubMed.
- J. M. Shi and C. W. Tang, Appl. Phys. Lett., 2002, 80, 3201 CrossRef CAS.
- C. Ego, D. Marsitzky, S. Becker, J. Zhang, A. C. Grimsdale, K. Müllen, J. D. MacKenzie, C. Silva and R. H. Friend, J. Am. Chem. Soc., 2003, 125, 437 CrossRef CAS.
- X. W. Chen, J. L. Liao, Y. M. Liang, M. O. Ahmed, H. E. Tseng and S.-A. Chen, J. Am. Chem. Soc., 2003, 125, 636 CrossRef CAS.
- J. X. Jiang, C. Y. Jiang, W. Yang, H. Y. Zhen, F. Huang and Y. Cao, Macromolecules, 2005, 38, 4072 CrossRef CAS.
- J. Liu, C. C. Min, Q. G. Zhou, Y. X. Cheng, L. X. Wang, D. G. Ma, X. B. Jing and F. S. Wang, Appl. Phys. Lett. Search PubMed , in revision.
- J. Liu, Q. G. Zhou, Y. X. Cheng, Y. H. Geng, L. X. Wang, D. G. Ma, X. B. Jing and F. S. Wang, Adv. Mater., 2005, 17, 2974 CrossRef CAS.
- G. L. Tu, Q. G. Zhou, Y. X. Cheng, L. X. Wang, D. G. Ma, X. B. Jing and F. S. Wang, Appl. Phys. Lett., 2004, 85, 2172 CrossRef CAS.
- G. L. Tu, C. Y. Mei, Q. G. Zhou, Y. X. Cheng, Y. H. Geng, L. X. Wang, D. G. Ma, X. B. Jing and F. S. Wang, Adv. Funct. Mater., 2006, 16, 101 CrossRef.
- Y. Q. Liu, G. Yu, H. Y. Liu, H. Tian and D. B. Zhu, Thin Solid Films, 2002, 417, 107 CrossRef CAS.
- F. Cacialli, R. H. Friend, C.-M. Bouché, P. L. Barny, H. Facoetti, F. Soyer and P. Robin, J. Appl. Phys., 1998, 83, 2343 CrossRef CAS.
- T. Förster, Discuss. Faraday Soc., 1959, 7, 27 Search PubMed.
- H. Suzuki and S. Hoshino, J. Appl. Phys., 1996, 79, 8816 CrossRef CAS.
- K. Utsugi and S. Takano, J. Electrochem. Soc., 1992, 139, 3610 CAS.
- S. Tasch, E. J. W. List, C. Hochfilzer, G. Leising, P. Schlichting, U. Rohr, Y. Geerts, U. Scherf and K. Müllen, Phys. Rev. B, 1997, 56, 4479 CrossRef CAS.
- Y.-H. Niu, J. Huang and Y. Cao, Adv. Mater., 2003, 15, 807 CrossRef CAS.
- T. Virgili, D. G. Lidzey and D. D. C. Bradley, Adv. Mater., 2000, 12, 58 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
-
A. J. Bard and L. A. Faulkner, Electrochemical Methods—Fundamentals and Applications, Wiley, New York, 1984 Search PubMed.
- M. S. Wrighton, D. S. Ginley and D. L. Morse, J. Phys. Chem., 1974, 78, 2229 CrossRef CAS.
- A. W. Grice, D. D. C. Bradley, M. Y. Bernius, M. Inbasekaran, W. W. Wu and E. P. Woo, Appl. Phys. Lett., 1998, 73, 629 CrossRef CAS.
- T. Miteva, A. Meisel, W. Knoll, H.-G. Nothofer, U. Scherf, D. C. Müller, K. Meerholz, A. Yasuda and D. Neher, Adv. Mater., 2001, 13, 565 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2006 |
Click here to see how this site uses Cookies. View our privacy policy here.