Stable aqueous dispersions of graphitic nanoplatelets via the reduction of exfoliated graphite oxide in the presence of poly(sodium 4-styrenesulfonate)
Sasha
Stankovich
a,
Richard D.
Piner
a,
Xinqi
Chen
a,
Nianqiang
Wu
b,
SonBinh T.
Nguyen
*c and
Rodney S.
Ruoff
*a
aDepartment of Mechanical Engineering, Northwestern University, 2145 Sheridan Rd., Evanston, IL 60206-3133, USA. E-mail: r-ruoff@northwestern.edu; Fax: +1(847)491-3915; Tel: +1(847)467-6596
bKeck Interdisciplinary Surface Science Center, NUANCE, 2220 Campus Drive, #2036, Northwestern University, Evanston, IL 60208, USA. Fax: +1(847)491-5429; Tel: +1(847)491-5505
cDepartment of Chemistry, 2145 Sheridan Rd., Evanston, IL 60208-3133, USA. E-mail: stn@northwestern.edu; Fax: +1(847)491-7713; Tel: +1(847)467-3347
First published on 21st November 2005
Abstract
For the first time, stable aqueous dispersions of polymer-coated graphitic nanoplatelets can be prepared via an exfoliation/in-situ reduction of graphite oxide in the presence of poly(sodium 4-styrenesulfonate).
Graphite nanoplatelets have recently attracted considerable attention as a viable and inexpensive filler substitute for carbon nanotubes in nanocomposites,1 given the predicted excellent in-plane mechanical, structural, thermal, and electrical properties of graphite.2 As with carbon nanotubes, full utilization of graphite nanoplatelets in polymer nanocomposite applications will inevitably depend on the ability to achieve complete dispersion of the nano-filler component in the polymer matrix of choice. In this communication, we describe a method for preparing water-soluble polymer-coated graphitic nanoplatelets.
We prepare graphite nanoplatelets via the chemical reduction of exfoliated graphite oxide nanoplatelets. Graphite oxide is produced by the oxidative treatment of graphite. It still possesses a layered structure, but is much lighter in color than graphite due to the loss of electronic conjugation brought about during the oxidation. The basal planes of the graphene sheets in graphite oxide are decorated mostly with epoxide and hydroxyl groups, in addition to carbonyl and carboxyl groups, which are located at the edges.3 These oxygen functionalities alter the van der Waals interactions between the layers of graphite oxide and render them hydrophilic, thus facilitating their hydration and exfoliation in aqueous media. As a result, graphite oxide readily forms stable colloidal dispersions of thin graphite oxide sheets in water.4 From these stable dispersions, thin “graphitic” nanoplatelets can be obtained by chemical deoxygenation, e.g., removal of the oxygen functionalities with partial restoration of the aromatic graphene network.5 It is possible that even single graphite sheets (i.e., finite-sized graphene sheets) can be accessed via graphite oxide exfoliation and a subsequent solution-based chemical reduction. In practice, reduction of water-dispersed graphite oxide nanoplatelets results in a gradual decrease in their hydrophilic character, which eventually leads to their irreversible agglomeration and precipitation. However, stable aqueous dispersions of reduced graphite oxide nanoplatelets can be prepared if the reduction is carried out in the presence of an anionic polymer.
A stable water dispersion of graphite oxide nanoplatelets, prepared by exfoliation of the graphite oxide (1 mg mL−1)6via ultrasonic treatment (Fisher Scientific FS60, 1 h), was reduced with hydrazine hydrate7 at 100 °C for 24 h. As the reduction proceeds, the brown-colored dispersion of exfoliated graphite oxide turns black and the reduced nanoplatelets agglomerate and eventually precipitate. This precipitated material could not be re-suspended even after prolonged ultrasonic treatment in water in the presence of surfactants such as sodium dodecylsulfate (SDS) and TRITON X-100, which have been found to successfully solubilize carbon nanotubes.8 Elemental analyses, coupled with Karl Fisher titration9 (Galbraith Laboratories), of both graphite oxide and the reduced material indicate that there is a considerable increase in C/O atomic ratio in the reduced material (10.3) compared to that in the starting graphite oxide (2.7). Hence, the reduced material can be described as consisting of partially oxidized graphitic nanoplatelets, given that a fair amount of oxygen is retained even after reduction. The black color of the reduced materials suggests a partial re-graphitization of the exfoliated graphite oxide, as observed by others.5 In addition to the decrease in the oxygen level, reduction of graphite oxide is accompanied by nitrogen incorporation from the reducing agent (C/N = 16.1).
Attempts to reduce graphite oxide in the presence of SDS and TRITON-X100 also failed to produce a stable aqueous dispersion of graphitic nanoplatelets. However, when the reduction was carried out in the presence of poly(sodium 4-styrenesulfonate) (PSS) (Mw = 70000, Sigma-Aldrich, 10 mg mL−1, 10/1 w/w vs. graphite oxide),10 a stable black dispersion was obtained. This dispersion can be filtered through a PVDF membrane (0.2 µm pore size, Fisher Scientific) to yield PSS-coated graphitic nanoplatelets that can be re-dispersed readily in water upon mild sonication, forming black suspensions (Fig. 1). At concentrations lower than 0.1 mg mL−1, the dispersions obtained after a 30-minute ultrasonic treatment appear to be stable indefinitely—samples prepared over a year ago are still homogeneous to date. More concentrated dispersions would develop a small amount of precipitate after several days. However, they never fully settle, even upon months of standing. Elemental analysis of the PSS-coated platelets indicates that it contains ∼40% polymer as judged by its sulfur content (graphite oxide reduced without any PSS contains no sulfur at all). Its comparatively high oxygen and hydrogen contents are attributed to the water adsorbed by the hygroscopic polymer (12% by Karl Fisher titration).11 The C/O atomic ratio in the reduced platelets alone is thus 9.3 (C/N ratio = 22.1), suggesting that a significant reduction of exfoliated graphite oxide platelets did take place in the presence of the polymer.
 | ||
| Fig. 1 Picture showing water dispersions (1 mg mL−1) of reduced exfoliated graphite oxide without PSS (left) and with PSS (right). | ||
That a large concentration of PSS is required to prevent the agglomeration of the platelets during reduction while only a fraction of that amount (<5.8% by weight of the PSS employed is absorbed on the reduced graphite oxide)12 is needed to render the complex dispersible indicates that the self-association between reduced graphite oxide platelets is a more favorable process than their association with PSS. During the reduction, the polymer needs to be present in a sufficient concentration for its interaction with the surfaces of the platelets to effectively compete with the hydrophobic interaction between the platelets. However, once PSS is attached to the platelet surface, further agglomeration of the platelets is stopped. When reductions were carried out without any PSS, the platelet agglomeration was irreversible: sonication of the reduced material after agglomeration in the presence of excess PSS did not result in platelet dispersion to any extent.
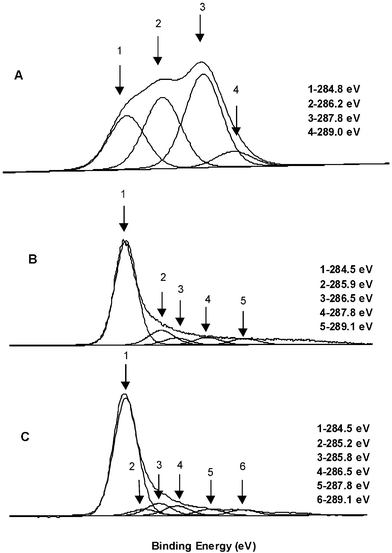
X-Ray photoelectron spectroscopy (XPS) (Omicron ESCA Probe, Omicron Nanotechnology) was employed to analyze the graphite oxide, the reduced graphite oxide, and the PSS-coated reduced graphite oxide. The C 1s XPS spectrum of graphite oxide (Fig. 2a) clearly indicates a considerable degree of oxidation with four components that correspond to carbon atoms in different functional groups:13 the non-oxygenated ring C (284.8 eV), the C in C–O bonds (286.2 eV), the carbonyl C (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 287.8 eV), and the carboxylate carbon (O–CO, 289.0 eV).14 Although the C 1s XPS spectra of the reduced graphite oxide (Fig. 2b) and the PSS-coated reduced graphite oxide (Fig. 2c) also exhibit the same oxygen functionalities that have been assigned for graphite oxide, the peak intensities of these components in the reduced samples are much smaller than those in the graphite oxide, indicating considerable de-oxygenation by the reduction process. In addition, there is an additional component at 285.9 eV corresponding to the C in the CN bonds of hydrazones.15
O, 287.8 eV), and the carboxylate carbon (O–CO, 289.0 eV).14 Although the C 1s XPS spectra of the reduced graphite oxide (Fig. 2b) and the PSS-coated reduced graphite oxide (Fig. 2c) also exhibit the same oxygen functionalities that have been assigned for graphite oxide, the peak intensities of these components in the reduced samples are much smaller than those in the graphite oxide, indicating considerable de-oxygenation by the reduction process. In addition, there is an additional component at 285.9 eV corresponding to the C in the CN bonds of hydrazones.15
 | ||
| Fig. 2 The C 1s XPS spectra of: A) graphite oxide, B) reduced graphite oxide, C) PSS-coated reduced graphite oxide. | ||
The C 1s XPS spectrum of the PSS-coated reduced graphite oxide sample also contains a component peak at 285.2 eV corresponding to the sulfur-bonded carbon atoms from PSS. The XPS survey scan of the PSS-coated reduced graphite oxide sample shows its characteristic peaks at 167.3 eV (S 2p, corresponding to sulfonate), 1071.7 eV (Na 1s), and 495.9 eV (Na KKL). These peaks are entirely absent from the XPS survey scan of the reduced graphite oxide.
Analysis of the C 1s XPS peak intensities indicates a higher level of oxygen for both graphite oxide and each of the reduced samples than those obtained by the elemental analysis of the bulk material. For example, the areas of the four C 1s components of graphite oxide indicate that only 21% of all graphite oxide carbons are unfunctionalized, while for reduced graphite oxide and the PSS-coated reduced graphite oxide the corresponding values are 75% and 74%. These numbers suggest higher C/O and C/N atomic ratios than the ratios found by elemental analyses (vide supra). In addition, analysis of the XPS spectrum of PSS-coated reduced graphite oxide yields a lower sulfur content (C/S = ∼36) as compared to bulk elemental analysis (C/S = 20.5). Since XPS is a surface-sensitive analytical technique, the disagreement with elemental analysis could arise from a higher degree of oxygenation of the surface of these materials and surface adsorption of various contaminants. Nevertheless, the C 1s XPS spectra clearly show that the reduction of graphite oxide, with (Fig. 2b) and without the polymer present (Fig. 2c), resulted in the same oxygen functionalities (vide supra).
Based on previous studies of the structure of graphite oxide and its chemical reactivity and on our own analytical data, one can speculate that our procedures for reducing graphite oxide and producing polymer-coated reduced graphite oxide most likely yield partially reduced graphite oxide materials that are comprised of graphitic regions in addition to islands of oxygen- or nitrogen-functionalized carbon atoms.
The stable dispersions of polymer-coated platelets were also analyzed by UV-Vis spectroscopy. The UV-Vis spectrum of the PSS-coated platelet dispersion in water possesses similar features as that of PSS itself (Fig. 3). In the spectrum of the colorless filtrate, obtained by filtration of the PSS-coated platelet dispersion through a PTFE syringe membrane filter (0.45 µm pore size, Fisher Scientific), the intensity of the signal arising from the polymer is very small,16 indicating that the polymer is strongly attached to the platelet surface (Fig. 3).
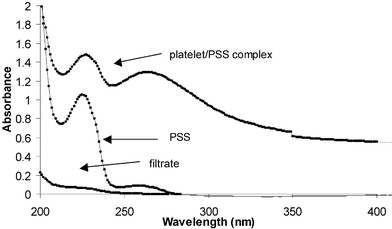 | ||
| Fig. 3 The UV-Vis spectra of the PSS-coated reduced graphite oxide complex (0.03 mg mL−1), PSS (0.025 mg mL−1), and the filtrate obtained by passing the platelet/polymer dispersion through a 0.45 µm PTFE membrane filter. | ||
While the IR spectrum of graphite oxide (GO) was quite informative,13 the FT-IR spectra of reduced GO and PSS-coated nanoplatelets were dominated by a broad background, making it difficult to identify the surface functionalities of the graphitic component. Independent of sample preparation techniques, the FT-IR spectra of reduced GO were essentially featureless, presumably due to the black color of this material. Although the FT-IR spectrum of PSS-coated nanoplatelets exhibits weak PSS absoprtion features, they only confirm the presence of the PSS component.
The visible (532 nm) Raman spectra of our samples were similarly uninformative. The only prominent features for all three samples—GO, reduced GO, and PSS-coated nanoplatelets—were the well-known D and G bands for the graphitic components.17 As PSS itself does not exhibits any Raman bands in our hands, it does not contribute to the Raman spectra of the PSS-coated nanoplatelets.
We analyzed the stable colloidal dispersions of exfoliated graphite oxide and PSS-coated reduced graphite oxide by atomic force microscopy (AFM) (AutoProbe CP/MT Scanning Probe Microscope (MultiTask), Veeco Instruments). AFM samples were prepared by depositing these dispersions on mica surfaces and allowing them to air-dry. The AFM image of the exfoliated graphite oxide revealed nanoplatelets with uniform thickness (∼1 nm) (such as shown in Fig. 4a). In the case of PSS-coated reduced graphite oxide, nanoplatelets with thickness of ∼4 nm were observed (Fig. 4b).
 | ||
| Fig. 4 A) Non-contact mode AFM image of exfoliated graphite oxide nanoplatelets (top). B) Non-contact mode AFM image of polymer-coated reduced graphite oxide nanoplatelets (bottom). | ||
Limited scale calculations of the interaction between PSS oligomers (degree of polymerization, DP = 6) and pristine graphene surface were carried out using MacroModel software (Schrodinger). The calculations indicate that edge-to-face interactions18 between the graphitic surface and the aromatic rings of the polymer might be primarily responsible for the polymer binding to the nanoplatelets, giving a maximum thickness for this interaction to be about 0.7 nm. From elemental analysis data, one polymer chain (Mw = 70 KDa, DP = 340) is responsible for coating roughly 8520 graphitic carbons, assuming also that both sides of the platelet are covered with the polymer and that each platelet consists of a single graphene (the single-sheet basic structural unit of graphite) sheet. This number corresponds to a 222 nm2 area of the graphene surface (with 0.026 nm2 as the cross-sectional area of a single graphene carbon, based on the 0.142 nm C–C bond length in graphite). Using 0.25 nm as the maximum monomer length for styrenyl monomers in the fully-extended all-trans configuration and 340 nm as the average number of monomer units in the PSS chain (vide supra), a simple calculation yields 85 nm for the average length of a fully extended PSS chain. Taking into account the length of the aromatic side groups (estimated at 0.8 nm from MacroModel calculations) and assuming that these groups project alternately from both sides of the polymer chain, the area occupied by a fully extended single polymer chain is 136 nm2. Although this estimate does not take into account the hydrated sodium ions that would certainly contribute to the orientation of the polymer chains on the graphene surface as well as the thickness of the polymer layer, it is consistent with a model where the partially reduced graphite oxide platelet surface is not fully graphitic and therefore is only partially covered with the polymer. Of course, we cannot rule out the possibility that the platelets are not uniformly single graphene sheets.
In conclusion, stable aqueous dispersions of graphitic nanoplatelets were prepared by coating reduced graphite oxide nanoplatelets with an amphiphilic polymer, poly(sodium 4-styrenesulfonate). To the best of our knowledge, this is the first example of stable dispersions of graphitic nanoplatelets in any solvent. The approach described in this work can be extended to obtain stable dispersion of graphitic nanoplatelets in organic solvents as well. Further work to achieve this goal is under way.
Acknowledgements
Support from NASA (Award # NCC-1-02037) through the University Research, Engineering, and Technology Institute on Bio-inspired Materials (BiMat) is appreciated. We thank Professor Jovica Badjic (Ohio State University) for performing the MacroModel calculation and for helpful discussions.Notes and references
- H. Fukushima and L. T. Drzal, Annu. Tech. Conf. Soc. Plast. Eng., 2003, 61, 2230–2234 Search PubMed.
- B. T. Kelly, Physics of Graphite, Applied Science, London, 1981, 475 pp Search PubMed.
- (a) H. He, T. Riedl, A. Lerf and J. Klinowski, J. Phys. Chem., 1996, 100, 19954–19958 CrossRef CAS; (b) H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS; (c) A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS; (d) C. Hontoria-Lucas, A. J. Lopez-Peinado, J. d. D. Loepz-Gonzalez, M. L. Rojas-Cervantes and R. M. Martin-Aranda, Carbon, 1995, 33, 1585–1592 CrossRef CAS.
- (a) M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929–2937 CrossRef CAS; (b) M. Hirata, T. Gotou and M. Ohba, Carbon, 2005, 43, 503–510 CrossRef CAS; (c) G. I. Titelman, V. Gelman, S. Bron, R. L. Khalfin, Y. Cohen and H. Bianco-Peled, Carbon, 2005, 43, 641–649 CrossRef CAS.
- (a) A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabo, A. Szeri and I. Dekany, Langmuir, 2003, 19, 6050–6055 CrossRef CAS; (b) U. Hofmann and A. Frenzel, Kolloid Z., 1934, 68, 149–151 CrossRef CAS.
- Graphite oxide was prepared from SP1 graphite (Bay Carbon, Bay City, MI) using a literature procedure. See: W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 Search PubMed.
- (a) P. Liu and K. Gong, Carbon, 1999, 37, 706–707 CrossRef CAS; (b) U. Hofmann and E. Konig, Z. Anorg. Allg. Chem., 1937, 234, 311–336 CrossRef CAS.
- V. C. Moore, M. S. Strano, E. H. Haroz, R. H. Hauge, R. E. Smalley, J. Schmidt and Y. Talmon, Nano Lett., 2003, 3, 1379–1382 CrossRef CAS.
- Water content analysis was necessary as graphite oxide is a hygroscopic material and the water trapped between its layers certainly contributes to the higher oxygen content (water content as high as 25% was measured in our graphite oxide samples). The water content in the reduced material is much lower, as expected, and was measured to be 2.79%.
- Although the polymeric surfactant is expected to bind more strongly to the surface of the nanoplatelets in comparison to the monomeric surfactants, a high concentration of it is still needed to prevent precipitation.
- Even drying of the material at 150 °C and 3 mTorr for 24 h failed to alter its elemental composition.
- Based on elemental analysis and the assumption that the polymer nanoplatelet complex is quantitatively isolated.
- The presence of these functional groups is further confirmed by the FT-IR spectrum (obtained with a Nexus 870 spectrometer, Thermo Nicolet) of graphite oxide. The most prominent features in the spectrum are the adsorption bands corresponding to the O–H stretching vibrations at 3428 cm−1, the CO carbonyl stretching at 1733 cm−1, the phenol CC ring stretching at 1621 cm−1, the phenol O–H deformation vibration at 1412 cm−1, the C–O stretching at 1053 cm−1, and the C–OH stretching, presumably from phenols, at 1226 cm−1.
- D. Briggs and G. Beamson, High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, John Wiley & Sons Inc, New York, 1992, 266 pp Search PubMed.
- It is likely that hydrazone formation occurs through the reaction of hydrazine with the carbonyl groups of the graphite oxide. The N 1s region in the XPS spectra of these reduced graphite oxide samples also exhibits two N peaks. See: R. J. Waltman, J. Pacansky and C. W. Bates, Jr., Chem. Mater., 1993, 5, 1799–1804 Search PubMed.
- Small amounts of PSS in the filtrate might be the result of incomplete removal of the excess polymer during the washing step in the synthesis of the polymer–platelet complex. They might also have been released from the platelet surface during the ultrasonic treatment needed to redisperse the dry polymer–platelet complex into water.
- J. Robertson, in Graphite and Precursors, ed. P. Delhaès, Gordon and Breach Publishers, Amsterdam, 2001, p. 257 Search PubMed.
- C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651–669 RSC.
| This journal is © The Royal Society of Chemistry 2006 |