DOI:
10.1039/B511349K
(Paper)
J. Mater. Chem., 2006,
16, 83-89
Synthesis and properties of conjugated oligomers containing fluorene, fluorenone, thiophene and cyclopentadithiophenone units
Received
9th August 2005
, Accepted 15th September 2005
First published on 5th October 2005
Abstract
Conjugated oligomers based on fluorene, fluorenone and thiophene derivatives have been prepared by parallel synthesis using the Suzuki cross-coupling reaction. These oligomers can be used as models of the corresponding fluorene copolymers and their structures were correlated with the observed optical and electrochemical behaviour. The long wavelength absorption bands observed for oligomers that contain fluorenone or cyclopentadithiophene units were assigned to an allowed π–π* transition associated with the carbonyl containing monomer unit. In addition the photoluminescence efficiency and the concentration dependence of the long wavelength emission band (>525 nm) for these materials strongly suggests that it can be attributed to emission involving fluorenone-based excimers.
1. Introduction
Polyfluorene homopolymers and copolymers are promising materials for application in organic light emitting diodes as they show high fluorescence efficiencies in the solid state, can be readily processed from solution, form high quality thin films and emit in the desirable blue region of the spectrum. Unfortunately, it has been shown that these materials undergo irreversible changes in the emission spectra on device operation unless the starting monomers are rigorously purified to remove any fluorene units that carry a labile H atom in the 9-position.1 In these experiments the desirable blue emission of the pristine polymer is rapidly transformed to a lower energy blue–green emission.2,3,4 The nature of the electronic state responsible for this emission has been controversial and although it is now clear that it is associated with the formation of fluorenone units in the polymer backbone it has been previously assigned to emission from on-chain fluorenone units,1,4 excimers,3 aggregates5 and most recently and perhaps most convincingly to an excimer associated with a fluorenone unit.6 Most studies of this phenomena have examined the photophysical behaviour of oxidised polymers,1,7–9 random copolymers of fluorene and fluorenone units2,4,10–12 or blends of fluorene polymers with fluorenone.13,14 There have been few examples of the use of fluorene/fluorenone-containing oligomers as models of the backbone units associated with this blue–green emission. This approach is promising as previous reports have shown that fluorene oligomers act as excellent models of polyfluorenes and the dioctylfluorene–fluorenone–dioctyl fluorene oligomer (Fig. 1) can act as an effective model of the emissive properties of polydioctylfluorene–fluorenone blends.
 |
| | Fig. 1 Chemical structure of dioctylfluorene–fluorenone–dioctyl fluorene oligomer. | |
In this contribution the solution phase synthesis of a library of conjugated oligomers by parallel methods is described. The library contains combinations of fluorene, thiophene, fluorenone and cyclopentadithiophene units to generate oligomeric models for the important structural elements of conjugated polymers based on a polydioctylfluorene backbone.
2. Experimental
Monomers 1–8 were synthesised following standard literature procedures.15–18
1H and 13C NMR spectra were collected using Bruker AC 250 MHz and AMX 400 MHz spectrometers. CHN analysis was determined using a Perkin-Elmer 2400 CHN elemental analyser. Purification of the library was performed by automated preparative liquid chromatography by using a Varian Prostar chromatograph with a Phenomonex LUNA column (150 mm × 21.20 mm, 5μ), an isocratic 85 : 15 THF–water mixture as eluent, flow rate 10 mL min−1, injection volume 0.5 mL, UV detector and a fraction collector (Varian model 701). Analytical HPLC was performed in similar equipment, Phenomonex LUNA column (150 mm × 4.60 mm, 5μ), a gradient from 50 to 100 % of THF in water as eluent, flow rate 1 mL min−1 and UV detection mode. The masses of the resulting products were determined by MALDI-TOF mass spectrometry using a Bruker Reflex III MALDI-TOF mass spectrometer. UV–Vis measurements were obtained on a Varian Cary 5000 UV–vis–NIR spectrophotometer. Photoluminescence measurements were carried out on a Varian Cary Eclipse fluorescence spectrophotometer. Films were spin-coated from dichloromethane at 2500 rpm. Cyclic voltammetry was performed using an EG & G Princeton Applied Research potentiostat/galvanostat (Model 273A) with a three-electrode cell in a solution of 0.1 M tetrabutylammonium hexafluorophospate (Bu4NPF6) as a supporting electrolyte in dry dichloromethane at a scan rate of 50 mV s−1. Thermal properties were studied by differential scanning calorimetry on a Perkin-Elmer Pyris 1 analyser (scan rate 10 °C min−1 and temperature range from 25 °C to 250 °C), and by polarised optical microscopy on an Olympus BH2 microscope attached to a temperature controller module Linkam TMS 94.
General Suzuki cross-coupling protocol
Monomers 2–5 and 7 were added to an equivalent molar amount of the boronic ester 1, 6 or 8, calculated to give around 500 mg of product in a Radley's carousel tube. Pd(PPh3)4 (2 mol%) as catalyst was added and the tubes were degassed by repetitive alternate application of vacuum and nitrogen gas. The mixture was then dissolved in dry, degassed toluene and a nitrogen purged solution of Na2CO3 2M (base: monomer, 60 : 1; base: toluene, 1 : 1.5) was added. The reactions were heated at 90 °C for 9 h and worked up by initial removal of the solvent under low vacuum, dissolution in dichloromethane and then parallel washing/extraction/phase separation (using a IST VacMaster vacuum manifold and SPE-filter-syringes) with a dilute solution of HCl (ca. 1% v/v, 3 × 5 mL). Removal of the solvent under reduced pressure gave the crude products, 9–14. The pure products were finally isolated by preparative HPLC. Details of the synthesis and the characterisation data for each oligomer (9–14) are listed below.
9,9,9′,9′,9″,9″-Hexaoctyl-9H,9′H,9″H-[2,2′;7′2″]terfluorene (9)
Dibromofluorene 2 (218 mg, 0.40 mmol), boronic ester 1 (411 mg, 0.80 mmol), Pd(PPh3)4 (28 mg), toluene (7.2 mL), Na2CO3 2M (4.8 mL). 1H NMR (CDCl3): δ/ppm 7.77 (m, 6H), 7.64 (m, 8H), 7.36 (m, 6H), 2.04 (t, 12H), 1.11 (m, 60H), 0.81 (m, 30H). 13C NMR (CDCl3): δ/ppm 151.8, 151.5, 151.0, 140.8, 140.5, 140.3, 140.0, 127.0, 126.8, 126.1, 126.03, 122.9, 121.5, 119.9, 119.7, 55.3, 55.2, 40.4, 31.8, 30.0, 29.2, 23.8, 22.6, 14.0. Calculated for C87H122: C, 89.47; H, 10.53. Found: C, 89.48; H, 10.56; mp: 64.9 °C. Mass spectrum m/z: 1168.
5,5′-Bis-(9,9-dioctyl-9H-fluoren-2-yl)-[2,2′]bithiophene (10)
Dibromobithiophene 3 (161 mg, 0.47 mmol), boronic ester 1 (489 mg, 0.95 mmol), Pd(PPh3)4 (33 mg), toluene (8.5 mL), Na2CO3 2M (5.7 mL). 1H NMR (CDCl3): δ/ppm 7.62 (d, J = 7.94 Hz, 4H), 7.54 (s, 2H), 7.50 (d, J = 4.58 Hz, 2H), 7.26 (m, 8H), 7.15 (t, 2H), 1.93 (t, 8H), 0.99 (m, 40H), 0.73 (t, 12H), 0.60 (m, 8H). 13C NMR (CDCl3): δ/ppm 151.6, 150.9, 143.9, 140.9, 140.6, 136.4, 132.8, 127.2, 126.8, 124.4, 123.6, 122.9, 120.1, 119.7, 55.2, 40.4, 31.8, 30.0, 29.2, 23.7, 22.6, 14.1. Calculated for C66H86S2: C, 84.02; H, 9.19. Found: C, 83.90; H, 9.06; mp: 114.4 °C. Mass spectrum m/z: 943.
2,6-Bis-(9,9-dioctyl-9H-fluoren-2-yl)-cyclopenta[2,1-b;3,4-b′]dithiophen-4-one (11)
Dibromocyclopentadithiophenone 4 (165 mg, 0.47 mmol), boronic ester 1 (486 mg, 0.94 mmol), Pd(PPh3)4 (33 mg), toluene (8.5 mL), Na2CO3 2M (5.6 mL). 1H NMR (CDCl3): δ/ppm 7.69 (dd, J = 2.7 Hz, J = 2.14 Hz, 4H), 7.52 (d, J = 1.52 Hz, 2H), 7.49 (s, 2H), 7.33 (m, 6H), 7.29 (s, 2H), 1.98 (t, 8H), 1.06 (m, 40H), 0.81 (t, 12H), 0.66 (m, 8H). 13C NMR (CDCl3): δ/ppm 151.8, 150.9, 148.3, 147.6, 142.3, 141.4, 140.3, 132.4, 127.4, 126.9, 124.1, 122.9, 120.2, 119.8, 119.4, 117.0, 55.2, 40.3, 31.8, 30.0, 29.2, 23.8, 22.6, 14.1. Calculated for C67H84OS2: C, 83.00; H, 8.73. Found: C, 82.85; H, 8.71; mp: 149.5 °C. Mass spectrum m/z: 969.
9,9,9″,9″-Tetraoctyl-9H,9″H-[2,2′;7′,2″]terfluoren-9′-one (12)
Dibromofluorenone 5 (158 mg, 0.47 mmol), boronic ester 1 (483 mg, 0.93 mmol), Pd(PPh3)4 (32 mg), toluene (8.4 mL), Na2CO3 2M (5.6 mL). 1H NMR (CDCl3): δ/ppm 8.03 (s, 2H), 7.76 (m, 6H), 7.64 (m, 4H), 7.60 (s, 2H), 7.35 (m, 6H), 2.03 (t, 8H), 1.06 (m, 40H), 0.79 (t, 12H), 0.66 (m, 8). 13C NMR (CDCl3): δ/ppm 151.6, 151.0, 142.9, 142.7, 141.2, 140.5, 138.5, 135.3, 133.4, 127.3, 126.8, 125.6, 123.1, 122.9, 121.0, 120.7, 120.1, 119.8, 55.3, 40.4, 31.8, 30.0, 29.2, 23.8, 22.6, 14.0. Calculated for C71H88O: C, 89.06; H, 9.26. Found: C, 87.19; H, 9.23; mp: 86.9 °C. Mass spectrum m/z: 957.
9′,9′-Dioctyl-9′H-[2,2′;7′,2″]terfluorene-9,9″-dione (13)
Bromofluorenone 7 (228 mg, 0.44 mmol), boronic ester 6 (282 mg, 0.22 mmol), Pd(PPh3)4 (30 mg), toluene (7.9 mL), Na2CO3 2M (5.3 mL). 1H NMR (CDCl3): δ/ppm 8.01 (d, J = 1.52 Hz, 2H), 7.80 (m, 4H), 7.70 (d, J = 7.33 Hz, 2H), 7.62 (m, 10H), 7.31 (m, 2H), 2.60 (m, 4H), 1.10 (m, 20H), 0.76 (t, 6H), 0.68 (m, 4H). 13C NMR (CDCl3): δ/ppm 152.0 144.4, 142.7, 140.6, 138.8, 134.9, 134.5, 133.2, 129.0, 125.8, 124.5, 123.0, 121.1, 120.7, 120.4, 120.3, 118.9, 55.5, 40.5, 31.7, 30.0, 29.2, 23.8, 22.6, 14.0. Calculated for C55H54O2: C, 88.43; H, 7.29. Found: C, 87.88; H, 7.18; mp: 230.0 °C. Mass spectrum m/z: 747.
9′,9′,9″,9″-Tetraoctyl-9′H,9″H-[2,2′;7′,2″]terfluoren-9-one (14)
Bromofluorenone 7 (124 mg, 0.48 mmol), boronic ester 8 (434 mg, 0.48 mmol), Pd(PPh3)4 (17 mg), toluene (4.3 mL), Na2CO3 2M (2.9 mL). 1H NMR (CDCl3): δ/ppm 8.02 (d, J = 1.2 Hz, 1H), 7.69 (m, 15H), 7.33 (m, 4H), 2.07 (m, 8H), 1.09 (m, 40H), 0.79 (q, 20H). 13C NMR (CDCl3): δ/ppm 152.0, 151.8, 151.5, 151.0, 144.5, 143.1, 142.8, 140.8, 140.4, 139.7, 138.6, 134.9, 134.6, 133.3, 129.0, 127.0, 126.8, 126.1, 125.8, 124.5, 123.0, 121.5, 121.1, 120.8, 120.4, 120.1, 119.9, 119.8, 119.0, 68.0, 55.5, 55.2, 40.4, 31.8, 30.1, 29.3, 23.9, 22.6, 14.1. Calculated for C71H88O: C, 89.06; H, 9.26. Found: C, 88.19; H, 9.53; mp: 42.7 °C. Mass spectrum m/z: 956.
3. Results and discussion
The synthetic protocol to obtain the library of oligomers (9–14) is shown in Scheme 1. The materials were synthesised in parallel by standard Suzuki cross-coupling of the boronic esters 1, 6 and 8 and the corresponding mono- or di-brominated derivatives (2–5,7). Initial reaction work-up consisted of parallel filtration, extraction and phase separation. Analysis of the crude reaction mixtures by HPLC showed that all materials contained the desired products (75–91%) with small amounts of unreacted starting materials and partial coupling products. The compounds were purified by preparative HPLC (see Fig. 2 for an example and analytical data in the Experimental). MALDI-TOF mass spectrometry showed a molecular ion at the correct mass for the desired products and the structures were confirmed by NMR spectroscopy and elemental analysis (see Experimental).
 |
| | Scheme 1 Synthetic protocol to obtain the library of oligomers. All the Suzuki coupling were carried out by using Pd(PPh3)4 as catalyst, Na2CO3 as base and toluene as solvent (see experimental). R: Octyl. | |
 |
| | Fig. 2 Liquid chromatogram for 9 and the corresponding MALDI-TOF mass spectra showing excellent purity from preparative HPLC. | |
Recent reports19 have shown that monodisperse oligofluorenes may show nematic liquid crystal phases, similar to that observed for high molecular weight polyfluorenes. Oligomers 9–14 showed no evidence for the formation of liquid crystal phases when studied by DSC or polarised optical microscopy. For the fluorenone substituted materials, melting points were observed to decrease from 222 °C for 13 to 87 and 65 °C for 12 and 14, respectively. In oligomers 9–14 it appears that the presence of long alkyl chains and the low degree of oligomerisation disfavoured the formation of liquid crystalline phases.
The infra-red spectra of 11–14 (see Fig. 3) showed a characteristic band at 1721 cm−1 for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, that was absent in the spectrum of the parent trifluorene 9. This band is significantly more intense for the oligomer with two fluorenone units (13) than those with only one keto group, as expected. The absorption at 1450 cm−1 can be assigned to the vibration of the aromatic ring and that at 1605 cm−1 related to the presence of an asymmetrically substituted benzene.2 This absorption is significantly more intense for 12, 13 and 14, specifically the oligomers that contain fluorenone substituents.
O stretch, that was absent in the spectrum of the parent trifluorene 9. This band is significantly more intense for the oligomer with two fluorenone units (13) than those with only one keto group, as expected. The absorption at 1450 cm−1 can be assigned to the vibration of the aromatic ring and that at 1605 cm−1 related to the presence of an asymmetrically substituted benzene.2 This absorption is significantly more intense for 12, 13 and 14, specifically the oligomers that contain fluorenone substituents.
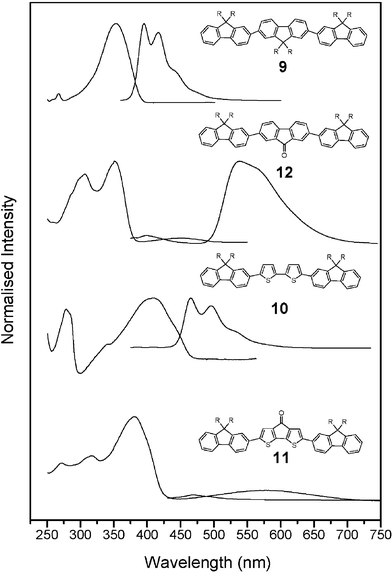
Given the number of recent reports describing the electroluminescence of fluorenone containing polymers,2,4,7,10–14 the optical properties of oligomers 11–14 are of interest. The absorption and emission maxima found in the UV–vis absorption and photoluminescence spectra are summarised in Table 1 and are shown in Figs. 4 and 5. Clearly, the structure of the oligomers strongly affects the optical properties. The maximum absorption (351 nm) of the parent dioctyl fluorene trimer 9 has been reported.20 Substitution of fluorene units for fluorenones in oligomers 12–14 results in little change in the position of the maximum absorption over that of the parent trimer and these values are comparable to that of the dihexylfluorene analogue of 12 (352 nm) previously reported.2 The maximum absorptions are all observed at longer wavelength than that of fluorenone (258 nm, ε 1.8 × 104 mol−1 dm3 cm−1, THF). As expected the absorption maximum for those oligomers that contain bithiophene and 4H-cyclopenta[2,1-b;3,4-b′]dithiophen-4-one units (10,11) are red-shifted from that of oligomer 9.
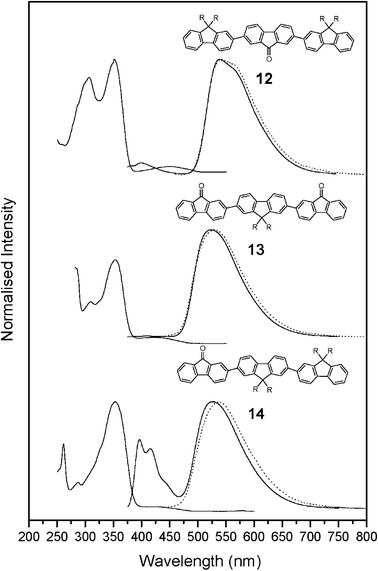 |
| | Fig. 5 UV–Vis absorption and photoluminescence spectra of fluorenone-substituted oligomers 12–14 in THF solution. Dotted lines indicate spectra recorded when the oligomer 12 was excited at 450 nm and when 13 and 14 were excited at 430 nm. | |
Table 1 Optical properties of the oligomers
| Oligomer |
Absorption |
Photoluminescence |
PL Efficiency(ΦPL)c |
| Solutiona |
Filmb |
Solutiona |
Filmb |
|
λ
max/nm |
λ
wk/nm |
λ
max/nm |
λ
wk/nm |
λ
max/nm |
λ
max/nm |
|
THF solution.
Spin-coated films.
Quantum yields of emission calculated by using quinine sulfate in 0.1 mol dm−3 H2SO4 (ΦPL = 0.546) as a standard and corrected for the refractive indices of solvents used,29,30 the values are estimated to ±0.01.
Extinction coefficients in mol−1 dm3 cm−1.
|
|
9
|
351 (155131)d |
— |
353 |
— |
395 |
424 |
0.90 |
|
10
|
407 (44302)d |
— |
399 |
— |
466 |
569 |
0.23 |
|
11
|
378 (33645)d |
577 (5299)d |
387 |
585 |
471 |
532 |
— |
|
12
|
350 (76594)d |
450 (3909)d |
361 |
451 |
540 |
552 |
0.11 |
|
13
|
351 (56164)d |
430 (5032)d |
361 |
442 |
528 |
531 |
0.16 |
|
14
|
351 (103764)d |
430 (5293)d |
357 |
431 |
525 |
525 |
0.15 |
The optical properties of fluorenone-containing polymers have been extensively discussed in the literature and remain controversial. It has been reported that an absorption band at 374 nm can be assigned to the π–π* transition of the fluorene units in these polymers and a weak band at 450 nm to the symmetry-forbidden n–π* transition2,4,12,21 or a π–π* transition of the fluorenone units.22–24 Previous photophysical studies have shown that the energies of these transitions are very similar and the nature of this optical transition can be interchanged depending on the solvent system employed for the measurement.22,23 The fluorenone substituted oligomers, 12–14, showed an absorption at around 350 nm (ε 104–105 mol−1 dm3 cm−1) that can be assigned to the π–π* transition of the fluorene units (see Table 1). In addition these oligomers (12–14) show a moderately weak band at 450 nm (ε 3909–5293 mol−1 dm3 cm−1). The 4H-cyclopenta[2,1-b;3,4-b′]dithiophen-4-one oligomer 11 shows a π–π* transition at 378 nm (ε 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 645 mol−1 dm3 cm−1) and a moderately weak absorption at 575 nm (ε 5299 mol−1 dm3 cm−1). There was no clear evidence for the assignment of the weak bands in these spectra (11–14) to an n–π* transition and the evidence supports the assignment of this absorption to a π–π* transition. Firstly, the observed extinction coefficients are larger than those expected for a typical n–π* transition (generally ε < 1000 mol−1 dm3 cm−1), furthermore a significant red shift is observed on increasing the polarity of the solvent used to record the spectrum of oligomers 11, 12–14 (for an example, see Fig. 6). If this absorption was associated with an n–π* transition then a blue shift in this absorption is expected on increasing the solvent polarity because of a reduction in the excited state dipole moment of the carbonyl group and as protic solvents can hydrogen bond to the oxygen lone pairs of the carbonyl group in the excited state to lower the energy of the non-bonding orbital.25–27 In contrast increasing the solvent polarity would be expected to red shift a π–π* transition as a polar solvent can lower the energy of the π* orbital. In addition the intensity of this absorption in the solid-state spectra is greatly enhanced over that expected for an n–π* transition and is in the range of 20–30% of the intensity of the maximum absorption (see Fig. 7). Finally, the large red shift (125 nm) in the long wavelength absorption of the cyclopentadithiophenone containing oligomer 11 over that of the analogous fluorenone containing oligomer 12 would not be expected for assignment of this absorption as an n–π* transition, associated primarily with the carbonyl group.
645 mol−1 dm3 cm−1) and a moderately weak absorption at 575 nm (ε 5299 mol−1 dm3 cm−1). There was no clear evidence for the assignment of the weak bands in these spectra (11–14) to an n–π* transition and the evidence supports the assignment of this absorption to a π–π* transition. Firstly, the observed extinction coefficients are larger than those expected for a typical n–π* transition (generally ε < 1000 mol−1 dm3 cm−1), furthermore a significant red shift is observed on increasing the polarity of the solvent used to record the spectrum of oligomers 11, 12–14 (for an example, see Fig. 6). If this absorption was associated with an n–π* transition then a blue shift in this absorption is expected on increasing the solvent polarity because of a reduction in the excited state dipole moment of the carbonyl group and as protic solvents can hydrogen bond to the oxygen lone pairs of the carbonyl group in the excited state to lower the energy of the non-bonding orbital.25–27 In contrast increasing the solvent polarity would be expected to red shift a π–π* transition as a polar solvent can lower the energy of the π* orbital. In addition the intensity of this absorption in the solid-state spectra is greatly enhanced over that expected for an n–π* transition and is in the range of 20–30% of the intensity of the maximum absorption (see Fig. 7). Finally, the large red shift (125 nm) in the long wavelength absorption of the cyclopentadithiophenone containing oligomer 11 over that of the analogous fluorenone containing oligomer 12 would not be expected for assignment of this absorption as an n–π* transition, associated primarily with the carbonyl group.
 |
| | Fig. 6 UV–Vis absorption of 10−6 mol dm−3 solutions of 12 in different solvents, the λ values in nm for the weak peak are: 440 (hexane), 450 (toluene), 457 (DCM), 451 (THF) and 457 (chloroform). | |
Oligomer 9 is strongly emissive in solution and the solid state, with principal peaks at 394 and 424 nm, respectively. The solution fluorescence spectra is sharper and better resolved (well-defined first two vibronic peaks, see Fig. 4) than the absorption spectra, suggesting that in the S1 state, there is an increase in the double bond character between the conjugated units (i.e., quinoidal form) and hence a more planar configuration20 (see below and Table 2). Quantum yield efficiencies (ΦPL) are summarised in Table 1 and the ΦPL value, absorption and photoluminescence maximum wavelengths for 9 agree well with those reported in the literature.20,28 In relation to the parent trifluorene, the fluorenone containing oligomers 12–14 are less efficient emitters, with photoluminescence efficiencies of 11, 16 and 15%, respectively. This decrease in efficiency is coupled with a significant red shift in the maximum of the emission to beyond 525 nm. This long-wavelength emission in fluorenone containing polymers and oligomers has been variously assigned to an excimer,3 a π–π* transition of the fluorenone unit1,4 and more recently to the emission of an excimer associated with the fluorenone unit.6 It is well known that excimer formation decreases dramatically the photoluminescence efficiency and low ΦPL values have been previously reported for other fluorenone-containing materials.2,4 Excitation of oligomers 12–14 at either the maximum absorption (350 nm) or the absorption assigned to the π–π* band (430 or 450 nm) of the fluorenone unit gave identical emission spectra (see Fig. 5) and the maximum in the solid state emission is close to that observed in solution (see Fig. 7). The effect of solution concentration on the fluorescence spectra of oligomers 12 and 14 was studied and Fig. 8 shows the fluorescence spectra measured at three separate concentrations with the intensity normalised to that of the emission of the band at 400 and 395 nm, respectively. The intensity of the emission at 552 nm for 12 and 525 nm for 14 decreases relative to that of the shorter wavelength emission on decreasing the solution concentration. This observation supports the assertion that the long wavelength emission in these compounds results from an excimer associated with the fluorenone unit, these results are in good agreement with those reported recently by Bradley et al.6 Emission from the 4H-cyclopenta[2,1-b;3,4-b′]dithiophen-4-one containing oligomer 11 is strongly quenched in solution and the solid state, similar quenching was observed for the parent cyclopentadithiophenone.
 |
| | Fig. 8 Normalised PL emission of 12 (a) and 14 (b) in THF at different concentrations under 350 nm excitation. | |
Table 2 HOMO/LUMO energy levels and band gaps determined from electrochemical measurements
| Oligomer |
First oxidation potential/V |
Second oxidation potential/V |
First reduction potential/V |
HOMO/eVa |
LUMO/eVa |
E
g/eV |
|
Values calculated from the equations HOMO = −(4.44 + EOX) and LUMO = −(4.44 + ERed). 31–33
(R): reversible.
(QR): quasi-reversible. Potential versus Ag/AgCl in dry DCM and referenced to the ferrocene/ferrocenium couple (0.44 V versus Ag/AgCl).
|
|
9
|
1.27 (R)b |
1.50 (R) |
−1.02 (QR)c |
−5.71 |
−3.42 |
2.29 |
|
10
|
1.00 (R) |
1.31 (R) |
−1.05 (QR) |
−5.44 |
−3.39 |
2.05 |
|
11
|
1.02 (R) |
1.34 (R) |
−1.11 (R) |
−5.46 |
−3.33 |
2.13 |
|
12
|
1.48 (QR) |
1.66 (QR) |
−1.25 (R) |
−5.92 |
−3.19 |
2.73 |
|
13
|
1.44 (QR) |
1.78 (QR) |
−1.31 (R) |
−5.88 |
−3.13 |
2.75 |
|
14
|
1.33 (QR) |
1.64 (QR) |
−1.29 (R) |
−5.77 |
−3.15 |
2.62 |
The synthesis of poly(9,9′-dioctylfluorene-alt-bithiophene) (F8T2) was first reported in 1998 by the Dow Chemical Company.34,35 F8T2 has been used as the semiconducting layer of organic field effect transistors36,37,38 and in organic light emitting devices,39 oligomer 10 represents a well-defined structural model of this polymer. The absorption maxima for the polymer in a chloroform solution is 452 nm and the emission 495 nm,39 as expected these values are red-shifted to those obtained for the oligomer 10 (407 and 466 nm). In the solid state the polymer absorbs at 458, 479 nm and emits light at 511, 537 and 577 nm, while oligomer 10 absorbs at 399 nm and emits at 537, 569 and 619 nm, with the maxima appearing at 569 nm. The red-shift for the latter presumably due to the better solid state packing observed for the oligomer. The solution photoluminescence efficiency (ΦPL in Table 1) is comparable for both the polymer (0.20) and the oligomer 10 (0.23). The reported values of the HOMO and LUMO for F8T2 thin films, determined by electrochemistry, are −5.41 and −2.48 eV, resulting in a calculated band gap of 2.93 eV.39 This value is higher than that calculated from the solution electrochemistry of oligomer 10 (2.05 eV, see below and Table 2). The HOMO value is similar for both oligomer 10 and the polymer, but the electron affinity of the polymer is significantly less than the value determined in solution for 10 (−3.39 eV). This is due to the presence of two fluorene units in the oligomer 10, whereas the alternating copolymer has one electron-rich bithiophene unit per fluorene.
All of the oligomers 9–14 showed two reversible or quasi-reversible oxidations and one reversible or quasi-reversible reduction in cyclic voltammetry in THF (10−3 mol dm−3) solution (see Fig. 9 and Table 2). Using the observed oxidation and reduction potentials the HOMO-LUMO energy levels and separation can be estimated and the values for oligomers 9–14 are collected in Table 2. From this data it is clear that substitution of a fluorene unit for a more electron-deficient fluorenone lowered the energy of the HOMO level and raised the energy of the LUMO for oligomers 12, 13 and 14 relative to those calculated for the parent trifluorene 9. The trends in the HOMO–LUMO separation calculated for 12–14 agree with those estimated from the difference between the maximum in the absorption and emission spectra (Figs. 4 and 5) previously described (see Table 1). The presence of a fluorenone in the middle of oligomer 12 has a comparable effect to that of two fluorenone units at the end of oligomer 13, as the solution electrochemistry results in very similar values for the calculated HOMO and LUMO of these molecules. The influence of the position of the fluorenone unit in the oligomer structure is shown by the difference in electrochemical behaviour between 12 and 14. Compound 14 is 0.15 V easier to oxidise than 12, presumably due to the asymmetry of 14 and the presence of a molecular dipole.
4. Conclusions
A library of well-defined conjugated oligomers of fluorene, fluorenone and thiophene derivatives has been prepared using parallel synthesis. The structure of these oligomers was correlated with their optical and electrochemical behaviour. It appears that the absorption band between 430–450 nm observed for the oligomers containing fluorenone units can be assigned to an allowed π–π* transition of the fluorenone, whereas the concentration dependence of the emission band at over 525 nm for these materials suggests that it can be attributed to excimer emission involving the fluorenone unit.
Acknowledgements
We would like to thank to the Colombian Institute for the Development of Science and Technology—COLCIENCIAS—for a graduate scholarship and the UK DTI for funding to support the Organic Materials Innovation Centre at the University of Manchester.
References
- E. J. W. List, R. Guentner, P. S. de Freitas and U. Scherf, Adv. Mater., 2002, 14, 374 CrossRef CAS.
- A. P. Kulkarni, X. Kong and S. A. Jenekhe, J. Phys. Chem. B, 2004, 108, 8689 CrossRef CAS.
- V. N. Bliznyuk, S. A. Carter, J. C. Scott, G. Klärner, R. D. Miller and D. C. Miller, Macromolecules, 1999, 32, 361 CrossRef CAS.
- E. Zojer, A. Pogantsch, E. Hennebicq, D. Beljonne, J.-L. Brédas, P. Scandiucci de Freitas, U. Scherf and E. J. W. List, J. Chem. Phys., 2002, 117, 6794 CrossRef CAS.
- C. Silva, D. M. Russell, A. S. Dhoot, L. M. Herz, C. Daniel, N. C. Greenham, A. C. Arias, S. Setayesh, K. Müllen and R. H. Friend, J. Phys.: Condens. Matter, 2002, 14, 9803 CrossRef CAS.
- M. Sims, D. D. C. Bradley, M. Ariu, M. Koeberg, A. Asimakis, M. Grell and D. G. Lidzey, Adv. Funct. Mater., 2004, 14, 765 CrossRef CAS.
- J.-I. Lee, G. Klaerner and R. D. Miller, Chem. Mater., 1999, 11, 1083 CrossRef CAS.
- J. M. Lupton, M. R. Craig and E. W. Meijer, Appl. Phys. Lett., 2002, 80, 4489 CrossRef CAS.
- L. Romaner, A. Pogantsch, P. Scandiucci de Freitas, U. Scherf, M. Gaal, E. Zojer and E. J. W. List, Adv. Funct. Mater., 2003, 13, 597 CrossRef CAS.
- S. Panozzo, J.-C. Vial, Y. Kervella and O. Stéphan, J. Appl. Phys., 2002, 92, 3495 CrossRef CAS.
- X. Gong, D. Moses, A. J. Heeger and S. Xiao, Synth. Met., 2004, 141, 17 CrossRef CAS.
- R. Demadrille, P. Rannou, J. Bleuse, J.-L. Oddou and A. Pron, Macromolecules, 2003, 36, 7045 CrossRef CAS.
- M. Grell, D. D. C. Bradley, G. Ungar, J. Hill and K. S. Whitehead, Macromolecules, 1999, 32, 5810 CrossRef CAS.
- A. P. Kulkarni and S. A Jenekhe, Macromolecules, 2003, 36, 5285 CrossRef CAS.
- J. Ding, M. Day, G. Robertson and J. Roovers, Macromolecules, 2002, 35, 3474 CrossRef CAS.
- M. Ranger, D. Rondeau and M. Leclerc, Macromolecules, 1997, 30, 7686 CrossRef CAS.
- J. Z. Brzezinski and J. R. Reynolds, Synthesis, 2002, 1053 CrossRef CAS.
- M. Ranger and M. Leclerc, Macromolecules, 1999, 32, 3306 CrossRef CAS.
- Y. Geng, S. W. Culligan, A. Trajkovska, J. U. Wallace and S. H. Chen, Chem. Mater., 2003, 15, 542 CrossRef CAS.
- M. Belletê, M. Ranger, S. Beaupré, M. Leclerc and G. Durocher, Chem. Phys. Lett., 2000, 316, 101 CrossRef.
- F. Uckert, S. Setayesh and K. Müllen, Macromolecules, 1999, 32, 4519 CrossRef CAS.
- T. Kobayashi and S. Nagakura, Chem. Phys. Lett., 1976, 43, 429 CrossRef CAS.
- J. R. Heldt, J. Heldt, M. Józefowicz and J. Kaminski, J. Fluoresc., 2001, 11, 65 CrossRef CAS.
- K. Yoshihara and D. R. Kearns, J. Chem. Phys., 1966, 45, 1991 CrossRef CAS.
- G. J. Brealey and M. Kasha, J. Am. Chem. Soc., 1955, 77, 4462 CrossRef CAS.
- H. McConnell, J. Chem. Phys., 1952, 20, 700 CAS.
-
C. Reichardt, Solvents and Solvents Effects in Organic Chemistry; E-mail: VCH, New York, 1990 Search PubMed.
- S. Tirapattur, M. Belletête, N. Drolet, M. Leclerc and G. Durocher, Macromolecules, 2002, 35, 8889 CrossRef CAS.
- E. A. Travnicek and J. H. Webber, J. Phys. Chem., 1961, 65, 225.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067 RSC.
- H. Meng, J. Zheng, A. J. Lovinger, B.-C. Wang, P. G. Van Patten and Z. Bao, Chem. Mater., 2003, 15, 1778 CrossRef CAS.
- W. Yang, Q. Hou, C. Liu, Y. Niu, J. Huang, R. Yang and Y. Cao, J. Mater. Chem., 2003, 13, 1351 RSC.
- T. Johansson, W. Mammo, M. Svensson, M. R. Andersson and O. Inganäs, J. Mater. Chem., 2003, 13, 1316 RSC.
-
M. Inbasekaran, W. Wu and E. P. Woo, US Pat., 5777070, 1998 Search PubMed.
- M. T. Bernius, M. Inbasekaran, J. O'Brien and W. Wu, Adv. Mater., 2000, 12, 1737 CrossRef CAS.
- H. Sirringhaus, R. J. Wilson, R. H. Friend, M. Inbasekaran, W. Wu, E. P. Woo, M. Grell and D. D. C. Bradley, Appl. Phys. Lett., 2000, 77, 406 CrossRef CAS.
- L. Kinder, J. Kanicki and P. Petroff, Synth. Met., 2004, 146, 181 CrossRef CAS.
- E. Lim, B.-J. Jung, J. Lee, H.-K. Shim, J.-I. Lee, Y. S. Yang and L.-M. Do, Macromolecules, 2005, 38, 4531 CrossRef CAS.
- E. Lim, B.-J. Jung and H.-K. Shim, Macromolecules, 2003, 36, 4288 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2006 |
Click here to see how this site uses Cookies. View our privacy policy here.