Revisiting silica based ordered mesoporous materials: medical applications
María
Vallet-Regí
*a,
Luisa
Ruiz-González
b,
Isabel
Izquierdo-Barba
a and
José M.
González-Calbet
b
aDpto Química Inorgánica y Bioinorgánica, Fac. Farmacia UCM., Madrid, 28040, Spain. E-mail: vallet@farm.ucm.es; Fax: 91 394 1786; Tel: 91 394 1861
bDpto Química Inorgánica, Fac. Químicas UCM, Madrid, 28040, Spain. E-mail: jgcalbet@quim.ucm.es; Fax: 91 394 4352; Tel: 91 394 4342
First published on 12th October 2005
Abstract
The bioactivity behaviour of SBA-15, MCM-48, MCM-41 mesoporous materials, is revisited in this paper. The influence of their different textural and structural properties on apatite formation is outlined and strategies to modify their kinetics are proven and discussed. On the basis of the experimental data showing the feasibility of control of the bioactivity kinetics on mesoporous materials, together with their controlled drug release abilities, new possibilities for tissue engineering developments are proposed.
 María Vallet-Regí | María Vallet-Regí was born in Las Palmas, Spain, in 1946. She studied chemistry at the Universidad Complutense de Madrid (UCM) and received her PhD at the same University in 1974. She is a full Professor of inorganic chemistry and Head of the Department of Inorganic Chemistry and Bioinorganic at the Faculty of Pharmacy (UCM). Her current research field is solid state chemistry, covering aspects of synthesis, characterisation and reactivity in oxides and bioceramics. |
 Luisa Ruiz-González | Luisa Ruiz-González studied for her B.S. (1996) and PhD (2002) in chemistry at the Universidad Complutense de Madrid (UCM). She enjoyed a post-doc position at the Oxford University working in the characterization of carbon nanotubes. Her research interest includes synthesis and structural characterization by SAED and HREM techniques of perovskite related, mesoporous and nano materials. She combines research with educational tasks as an assistant at UCM. |
 Isabel Izquierdo-Barba | I. Izquierdo received her B.S. (1997) and PhD (2003) in pharmacy at the Universidad Complutense de Madrid (UCM). She enjoys a post-doc position at the Stockholm University working in the characterization of mesoporous materials in the Structural Chemistry department. |
 José M. González-Calbet | José M. González-Calbet was born in Madrid, Spain, in 1952. He studied chemistry at the Universidad Complutense de Madrid (UCM) and received his PhD at the same University in 1979. He is a full Professor of inorganic chemistry and Head of the Department of Inorganic Chemistry at the Faculty of Chemistry (UCM). His current research field is solid state chemistry, covering aspects of reactivity of oxides and structural characterisation by electron microscopy. |
The discovery of ordered silica-based mesoporous materials, by scientists of the Mobil Corporation1 in 1992, has attracted enormous interest allowing expansion of the potential applications of the host–guest system at the nanometre scale. These applications comprise a broad range of topics, from catalysis to controlled drug release. Actually, mesoporous materials constitute a new generation of materials that show ordered arrangements of channels and cavities of different geometry built up from SiO2 unities. The pore size is variable (2nm < Φp < 50 nm) and can be controlled and modified, in a reasonable range, using in situ2 and ex situ3 synthetic strategies. Well-known examples are the 2D-hexagonal (S.G. p6mm) MCM-414 and SBA-155 silica mesoporous materials with pores around 2 and 10 nm, respectively, and the 3D-cubic MCM-486 (S.G. Ia3d ) with pore size between 2 and 4 nm. The feasibility to obtain different pore size and geometries offers a wide range of possibilities for hosting molecules larger than the ones exhibited for classic microporous materials, such as zeolites. Moreover, the absorption properties can be modified, since the pore walls, exhibiting high concentration of silanol groups at the surface, can be functionalized with different chemical species. Taking into account these characteristics, the behaviour of mesoporous materials as drug delivery systems has been developed by this research group since 2001. It is based on the ability of these matrixes to absorb molecules, of pharmacological interest, followed by a potentially controlled release.7 This work has opened new directions on the medical research in drug release from a mesoporous matrix.7–17 Exploring new applications of mesoporous materials is a very attractive goal and, based on their composition and structure, it is possible to consider them as scaffolds in regeneration of hard tissues (bones and tooth). For such a desirable application, the mesoporous materials must exhibit a bioactive behaviour. At this point, it is worth recalling that bioactive glasses,18–24 that also exhibit silanol groups (Fig. 1), are able to develop an apatite layer. Since Hench et al. discovered Bioglass® in 197125 many studies, comprising different glasses and calcium silicates compositions, have shown in vitro bioactivity.22,26–29 Although the mechanism of apatite formation is not completely elucidated, the presence of silanol groups in the surface seems to be crucial. In this sense, several authors propose that silanol groups act as nucleation sites (Fig. 2). However, there are more factors compulsory to the apatite formation;30,31 among them, it is interesting to point out the textural properties, i.e. those related with the porosity. It has been proved that a direct relation between both pore size and volume and nucleation rate of the apatite layer32–34 exists.
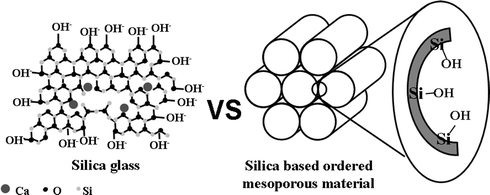 | ||
| Fig. 1 Schematic illustration of bioactive glass and ordered mesoporous materials, which shows that both materials exhibit high silanol concentration. | ||
 | ||
| Fig. 2 Hydroxyapatite formation mechanism in bioactive glasses when soaked in physiological fluids. | ||
Taking into account the above considerations, it seems logical to focus on mesoporous silica, because they show high specific surface, high concentration of silanol groups, feasible pore size (2–10 nm) and volume,35–38 and can also exhibit a bioactive behaviour. In this sense, bioactivity essays were performed in three well known mesoporous materials, SBA-15, MCM-48 and MCM-41. A positive response was found for SBA-15 and MCM-48.39 Although pure MCM-41 is not bioactive, it has been shown that minimal compositional changes can modify this behaviour.40,41 The aim of this work is to review the potential bioactive behaviour of three matrices in order to analyze the different observed responses, for further applications. In fact, the combination of the recent results obtained for SBA-15 and MCM-48, as well as the possibility not only to promote but also to modify the kinetics of apatite formation in MCM-41, allow us to think about new potential applications in medical science and, more specifically, in biomaterials and tissue engineering. Moreover, development possibilities can be enhanced considering the ability of these materials to accept drugs and osteogenic substances for controlled release. The biocompatibility of these materials makes them appropriate for clinical applications if they are either bioactive or bioinert.
Bioactivity in silica based ordered mesoporous materials
Mesoporous materials with different structural and textural properties were chosen in order to evaluate their influence on the corresponding in vitro response. Bioactivity studies on SBA-15, MCM-48 and MCM-41 materials have been carried out.39 The assessments of in vitro bioactivity were performed in simulated body fluid (SBF),42 which has the composition and ionic concentration similar to human plasma, at 37 °C during different immersion periods (Fig. 3). Different characterization techniques (FTIR, SEM-EDS, TEM-ED-EDX) show that, after the in vitro tests, SBA-15 and MCM-48 surfaces are modified, as a consequence of the reaction of the materials with the fluid, giving rise to the appearance of a carbonate hydroxyapatite layer.39 The kinetics of each process is quite different; SBA-15 develops the apatite layer after 30 d while 60 d are required in the case of MCM-48. Nevertheless, after 60 d of immersion the apatite layer did not appear in MCM-41. SEM micrographs and EDS of samples SBA-15 and MCM-48 before and after 15, 30 and 60 d in SBF show that the surface has been covered with needle-type crystal aggregates forming spherical particles. The EDS analysis confirms the presence of Ca and P on the surface. In the case of MCM-41, the surface is not modified and Ca and P have not been detected (Fig. 4).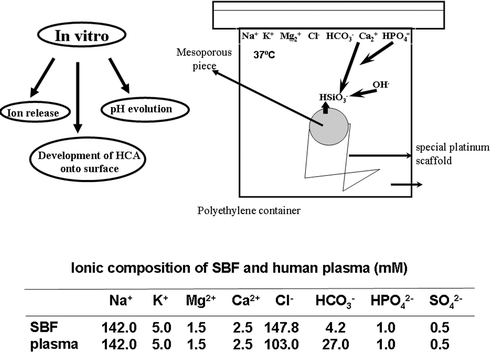 | ||
| Fig. 3 Schematic description of the in vitro bioactivity test. Ion concentration in SBF and in human blood plasma are also included. | ||
 | ||
| Fig. 4 SEM micrographs and EDS spectra of SBA-15, MCM-48 and MCM-41 materials at 0, 15, 30 and 60 d. | ||
A TEM study, before and after the treatment in SBF, was also performed (Fig. 5). HREM images before the treatment show the pore size characteristic of the raw material. After the indicated periods of time, the apatite formation is confirmed at SBA-15 and MCM-48 surfaces,39 as shown for SBA-15 in Fig. 5b and d. A careful inspection of the interface between the mesoporous material and the recently formed apatite supplies additional information (see Fig. 1), not previously available in bioactive glasses due to the lack of order. Crystalline particles embedded in an amorphous matrix are found, as clearly observed at the insets of Fig. 5. The amorphous area contains Si while the crystalline one comprises Ca and P. Since only Si is present at the amorphous matrix, it seems logical to relate it with the original mesoporous material that is transformed. This fact could be related to (i) the mesoporous surface modification as consequence of its reaction with the SBF to form the new apatite, the product of this chemical process or (ii) the transformation under the electron beam. A combination of these two factors is probably the more plausible explanation. Minor areas where the mesoporous material has not yet been completely decomposed are also found, as reflected in Fig. 6, corresponding to SBA-15. It is worth mentioning that in such cases the needle like particles are not placed over the mesoporous walls (Fig. 6a). On the left hand of the image, fringes of 8.8 nm periodicity, in agreement to SBA-15, in a clear process of modification, are observed. On the right hand, a small needle like particle is present. This is better seen in the enhanced image (Fig. 6b) where the crystalline nature of the particle imbibed in an amorphous area is also clearly observed. Measured periodicities correspond to the apatite cell. When the FT (Fourier transform) is performed on the amorphous area a broad diffuse scattering appears (Fig. 6c), reflecting, again, the amorphous nature. It is then clear that the mesoporous surface is modified when apatite is formed. This process cannot be studied on bioactive glasses due to their total structural disorder.
 | ||
| Fig. 5 TEM micrographs corresponding to SBA-15: (a) HREM image of the mesoporous material before treatment in SBF; (b) low magnification image after 60 d in SBF, the crystalline (inset A) and amorphous (inset B) nature of the particles and matrix, respectively, are clearly observed; (c) SAED pattern corresponding to the raw SBA-15 mesoporous material; (d) FT corresponding to inset A, characteristic of an apatite cell. | ||
 | ||
| Fig. 6 TEM micrographs showing different features of SBA-15 after treatment: (a) area where the original mesoporous material is not completely transformed; features of both raw material and apatite are evident; (b) enhanced image reflecting the fact that apatite appears over the modified mesoporous material; (c) FT performed at the amorphous area in b. | ||
The different behaviour shown for SBA-15, MCM-48 and MCM-41 can be understood on the basis of the compositional, textural and structural properties of the mesoporous materials, summarized in Table 1. As it has already been mentioned, the silanol groups (Si–OH) can act as nucleation sites of the apatite layer.22,26–31 In this sense, the silanol group concentration seems to be an important factor to consider. In this case, the Si–OH concentration per surface has been determined by thermogravimetric analyses (TGA) and N2 adsorption measurements. MCM-41 shows a rather lower concentration of silanol groups (ca. 2.0 mmol SiOH m−2) than SBA-15 and MCM-48 (ca. 13 mmol SiOH m−2). This could be one reason to explain the absence of bioactivity in MCM-41. Moreover, the textural and structural properties must also be taken into account in order to justify this negative behavior. Both factors are related with the possible ion diffusion inside the mesoporous material. Bigger and more accessible pores will favor the ion diffusion, i.e. the apatite nucleation will be faster.32–34 Contrarily, the ion diffusion becomes slower and difficult if the pore size decreases, diminishing the nucleation rate. In this sense, SBA-15 shows the largest pore size (8.8 nm) being considerably lower (3.6 nm) in the cases of MCM-48 and MCM-41. This fact could explain the better kinetics observed in SBA-15 compared to MCM-48 and MCM-41. Furthermore, the dimension and shape of the pores is also a very important factor to be considered. In this sense, MCM-41 has pure one dimensional character while MCM-48 and SBA-15 are connected through complementary pores favoring the diffusion paths. However, the identical pore size of MCM-48 and MCM-41 does not justify the great difference on their bioactive behavior, but can be understood taking into account the silanol concentration as well as the structure. MCM-41 wall silanol concentration is lower than in MCM-48 and the 2D-hexagonal lattice without channel connections is less accessible to the ions if compared with the 3D-cubic one exhibited by MCM-48. Moreover, local curvatures for MCM-41 comparing to SBA-15 and MCM-48, being both quite similar, are different and may also play an influence.
| Matrices | Bioactivity behaviour | Time in SBF/d | S BET/ m2 g−1 | D p/nm | V p/cm3 g−1 | V micropore/cm3 g−1 | [SiOH]/mmol m−2 |
|---|---|---|---|---|---|---|---|
| SBA-15 | Yes | 30 | 787 | 8.8 | 1.05 | 0.061 | 12.71 × 10−3 |
| MCM-48 | Yes | 60 | 1166 | 3.6 | 1.05 | — | 12.86 × 10−3 |
| MCM-41 | No | 60 | 1157 | 3.6 | 0.98 | — | 2.16 × 10−3 |
| P-MCM-41 | Yes | 15 | 721 | 3.6 | 0.91 | — | 2.93 × 10−3 |
| MCM-41/G | Yes | 1 | 1131 | 3.4 | 0.80 | — | 1.53 × 10−3 |
The above discussion allows us to understand, on the basis of compositional, textural and structural properties, why apatite formation happens in SBA-15 and MCM-48 in 30 and 60 d, respectively, while MCM-41 does not suffer any alteration, after two months of assay. This does not mean, in principle, that it is impossible to develop the apatite layer, but it is clear that the rate is too slow. Once that the factors playing an important role on the apatite formation have been discussed, the aim is to look for chemical procedures in order to modify the MCM-41 kinetics. On the basis of previous knowledge two different strategies have been applied: (i) doping with phosphorous (ii) addition of small amounts of a second substance that exhibits high bioactivity.
The ensemble of results concerning the five materials analysed in this study (Table 1) opens an enormous expectation about their possible applicability. The first hypothesis, on the basis of the evidence of bioactivity, is the possibility to design mesoporous materials in accordance with the desirable response time. In fact, MCM-41 does not develop apatite even after months of treatment, but it is attained after 15 and 1 d by doping with small amounts of P or adding bioactive glass, respectively. If the same procedures were applied to SBA-15 and MCM-48, that exhibit bioactivity after 30 and 60 d of treatment, respectively, it would be possible to adjust the kinetics of the apatite formation process to the required time of a specific problem.
Moreover, these materials can be functionalized with both polar and non polar molecules. This is very important from the biological point of view, since it opens up the possibility to attach different groups with affinity for cells or biological species (such as thiol, acid or amino) to the mesoporous wall. These groups can allow the attachment with specific proteins (osteogenic peptides and growth factors) opening new alternatives in tissue engineering. The loss or failure of an organ or tissue is one of the most frequent and expensive clinical problems. In this sense, regeneration constitutes a target objective of many studies. Tissue engineering is an emerging area directed towards the design of materials than can help an organism to improve its ability of regeneration by recovering both the structure and also its function. There are different strategies but one particularly interesting is the combination of cell harvest with scaffolds by means of in vitro techniques, followed by implantation on the corresponding organ of the host.50 For this purpose, biocompatible and bioresorbable scaffolds are desirable to favour the tissue growth and increase the cellular function, i.e., to help the development of cells into a functioning tissue, ready for implantation.
Taking into account these ideas and the mesoporous properties, here reviewed, it is clear that they are promising candidates as scaffolds for tissue regeneration (Fig. 7). For this purpose, it is necessary to find suitable combinations of bioactivity, drug release, porosity and wall functionalization.
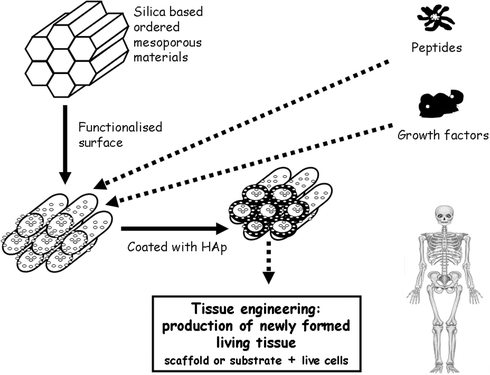 | ||
| Fig. 7 Schematic illustration of tissue engineering technique promoted by bioactive ordered mesoporous materials which can be functionalized with organic groups and release different growth factors for tissue regeneration. | ||
Conclusions
The role that the textural and structural properties of the mesoporous materials play on their bioactive behaviour is established. Alternatively, it is shown how the kinetics of the apatite formation can be modified and improved. The possibility to control the periods of time needed for a positive response, together with the ability to functionalise the surface and the introduction of osteogenic substances inside the pores, open new expectations for designing new mesoporous materials directed to specific medical applications.References
-
J. S. Beck, C. T.-W. Chu, I. D. Johnson, C. T. Kresge, M. E. Leonowicz, W. J. Roth and J.C. Vartuli, Mobil Oil, US Patent, 5
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 108725, 1992
108725, 1992 .
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS
- X. S. Zhao, G. Q. Lu and X. Hu, Chem. Commun., 1999, 1391 RSC
- A. Firouzi, F. Atef, A. G. Oertly, G. D. Stucky and B. Chmelka, J. Am. Chem. Soc., 1997, 119, 3596 CrossRef CAS
- D. Zhao, J. P. Feng, Q. S. Huo, N. Melosh, G. H. Fedrickson, B. F. Chemelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS
- M. Kaneda, T. Tsubakiyama, A. Carlsson, Y. Sakamoto, T. Ohsuna, O. Terasaki, S.-H. Joo and R. Ryoo, J. Phys. Chem. B, 2002, 103, 1256 CrossRef
- M. Vallet-Regí, A. Rámila, R. P. del Real and J. Pérez-Pariente, Chem. Mater., 2001, 13, 308 CrossRef CAS
- B. Muñoz, A. Rámila, I. Díaz, J. Pérez-Pariente and M. Vallet-Regí, Chem. Mater., 2003, 15, 500 CrossRef CAS
- A. Ramila, B. Munoz, J. Pérez-Pariente and M. Vallet-Regí, J. Sol–Gel Sci. Technol., 2003, 26, 1199 CrossRef CAS
- A. L. Doadrio, E. M. B. Sousa, J. C. Doadrio, J. Perez-Pariente, I. Izquierdo-Barba and M. Vallet-Regí, J. Controlled Release, 2004, 97, 125 CrossRef CAS
- M. Vallet-Regí, J. C. Doadrio, A. L. Doadrio, I. Izquierdo-Barba and J. Perez-Pariente, Solid State Ionics, 2004, 172, 435 CrossRef CAS
- C. Charnay, S. Bégu, C. Tourné-Péteilh, L. Nicole, D. A. Lerner and J. M. Devoisselle, Eur. J. Pharm. Biopharm., 2004, 57, 533 CrossRef CAS
- P. Horcajada, A. Rámila, J. Pérez-Pariente and M. Vallet-Regí, Microporous Mesoporous Mater., 2004, 68, 105 CrossRef CAS
- J. Anderson, J. Resenholm, S. Arev and M. Linden, Chem. Mater., 2004, 16, 4160 CrossRef
- B. G. Trewyn, C. M. Whitman and V. S.-Y. Lin, Nano Lett., 2004, 4, 2139 CrossRef CAS
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinifa, K. Jeftinifa, S. Xu, S. Jeftinifa and V. Lin, J. Am. Chem. Soc., 2003, 125, 4451 CrossRef CAS
- W. Zeng, X. F. Qian, Y. B. Zhang, J. Yin and Z. K. Zhu, Mater. Res. Bull., 2005, 40, 766 CrossRef CAS
- L. L. Hench and J. Wilson, Science, 1984, 226, 630 CrossRef CAS
- Ö. H. Andersson, K. H. Karlsson, K. Kangasniemi and A. Yli-Urpo, Glastech. Ber., 1988, 61, 300 Search PubMed
- T. Kokubo, J. Ceram. Soc. Jpn., 1991, 99, 965 CAS
- R. Li, A. E. Clark and L. L. Hench, J. Appl. Biomat., 1991, 2, 231 Search PubMed
- M. Vallet-Regí, J. Chem. Soc., Dalton Trans., 2001, 363, 254 Search PubMed
- H. M. Kim, J. Ceram. Soc. Jpn., 2001, 109, S49 CAS
- L. L. Hench and J. M. Pollack, Science, 2002, 295, 1014 CrossRef CAS
- L. L. Hench, R. J. Splinter, W. C. Allen and T. K. Greenlee, J. Biomed. Mater. Res., 1971, 2, 117 CrossRef
- M. Vallet-Regí, C. V. Ragel and J. A. Salinas, Eur. J. Inorg. Chem., 2003, 1029 CrossRef CAS
-
J. Black and G. Hastig, Handbook of Biomaterial Properties, Chapman and Hall, London, 1998 Search PubMed
-
Handbook of Bioactive Ceramics. Bioactive Glasses and Glass–ceramics, vol. 1, ed. T. Yamamuro, L. L. Hench, J. Wilson, CRC Press, Boca Raton, FL, 1990 Search PubMed
- M. Brink, T. Turunen, R. P. Happonen and A. Yli-Urpo, J. Biomed. Mater. Res., 1997, 37, 114 CrossRef CAS
- P. Li, C. Ohtsuki, T. Kokubo, K. Nakanishi, N. Soga, T. Nakamura and T. Yamamuro, J. Am. Ceram. Soc., 1992, 75, 2097
- S. B. Cho, K. Nakanishi, T. Kokubo and N. Soga, J. Am. Ceram. Soc., 1995, 78, 1769
- M. M. Pereira, A. E. Clark and L. L. Hench, J. Am. Ceram. Soc., 1995, 78, 2163
- M. M. Pereira and L. L. Hench, J. Sol–Gel Sci. Technol., 1996, 7, 59 CAS
- D. Arcos, D. C. Greenspan and M. Vallet-Regí, Chem. Mater., 2002, 14, 1515 CrossRef CAS
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartulli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS
- S. Inagaki, Y. Fukushima and K. Kuroda, J. Chem. Soc., Chem. Commun., 1993, 8, 680 RSC
- Q. Huo, D. I. Margolese, U. Ciesla, P. Y. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schuth and G. D. Stucky, Nature, 1994, 368, 317 CrossRef CAS
- Y. Sakamoto, M. Kaneda, O. Terasaki, D. Y. Zhao, J. M. Kim, G. Stucky, H. J. Shin and R. Ryoo, Nature, 2000, 408, 449 CrossRef CAS
- I. Izquierdo-Barba, L. Ruiz-González, J. C. Doadrio, J. M. González-Calbet and M. Vallet-Regí, Solid State Sci., 2005, 7, 983 CrossRef CAS
- M. Vallet-Regí, I. Izquierdo-Barba, A. Ramila, J. Pérez-Pariente, F. Babonneau and J. M. Gónzalez-Calbet, Solid State Sci., 2005, 7, 233 CrossRef CAS
- P. Horcajada, A. Ramila, K. Boulahya, J. M. Gonzalez-Calbet and M. Vallet-Regí, Solid State Sci., 2004, 6, 1295 CrossRef CAS
- T. Kokubo, H. Kushitani, S. Sakka, T. Kisugi and T. Yamamuro, J. Biomed. Mater. Res., 1990, 24, 721 CrossRef CAS
- M. Vallet-Regí, I. Izquierdo-Barba and A. J.Salinas, J. Biomed. Mater. Res., 1999, 46, 560 CrossRef CAS
- M. Vallet-Regí, A. Rámila, S. Padilla and B. Muñoz, J. Biomed. Mater. Res., 2003, 66, 580 CrossRef CAS
- I. M. O. Kangasniemi, E. Vedel, J. de Blick-Hogerworst, A. Yli-Urpo and K. de Groot, J. Biomed. Mater. Res., 1993, 27, 1225 CrossRef CAS
- J. D. Santos, J. J. Lakhan and F. J. Monteiro, Biomaterials, 1995, 16, 521 CrossRef CAS
- M. Vallet-Regí and A. Ramila, Chem. Mater., 2000, 12, 961 CrossRef CAS
- M. A. Lopes, J. D. Santos, F. J. Monteiro, C. Othsuki, A. Otaka, S. Kaneko and H. Inove, J. Biomed. Mater. Res., 2001, 54, 463 CrossRef CAS
- C. V Ragel, M. Vallet-Regí and L. M. Rodriguez-Lorenzo, Biomaterials, 2002, 23, 1865 CrossRef
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869 CrossRef
| This journal is © The Royal Society of Chemistry 2006 |