Speciation studies of cis-platin adducts with DNA nucleotides via elemental specific detection (P and Pt) using liquid chromatography-inductively coupled plasma-mass spectrometry and structural characterization by electrospray mass spectrometry†
Daniel Garcia Sar, Maria Montes-Bayón, Elisa Blanco González and Alfredo Sanz-Medel
Department of Physical and Analytical Chemistry, Faculty of Chemistry, University of Oviedo, C/Julián Clavería 8, 33006 Oviedo, Spain. E-mail: asm@uniovi.es
First published on 7th June 2006
Abstract
As the cis-platinum (cis-Pt) antitumoral effect in mammals seems to be related to its binding to DNA components, experiments with in vitro incubation of the individual DNA nucleotides with cis-Pt and analysis of the products by electrospray mass spectrometry (ESI-Q-TOF) are described here. The only detectable complex of such binding has been the one formed by a cis-Pt molecule bound to two adjacent guanines (m/z 921), as confirmed by collisional induced dissociation. The separation of the cis-Pt adducts from the unreacted nucleotides has been conducted by high-performance liquid chromatography coupled on-line with inductively coupled plasma mass spectrometry (HPLC-ICP-MS), monitoring 31P and 195Pt. Two different chromatographic columns have been evaluated for this purpose: a RP-amide-C16 and a narrow-bore C8. Best separation characteristics for the four nucleotides of DNA (coming from adenine, thymine, cytosine and guanine nucleobases) and the formed cis-Pt adduct were obtained for the C8 column using a mobile phase containing 60 mM ammonium acetate (pH = 5.8) and 7.5% MeOH. This HPLC-ICP-MS method allowed an easy separation and detection of free nucleotides (by monitoring P) from the synthesized adduct (containing P and Pt in the same molecule). Quantitative capabilities of the proposed hybrid method, by monitoring 31P and 195Pt, have been compared by analysing the cis-Pt adduct formed by the oligonucleotide of sequence 5′-TCCGGTCC-3′ after incubation with cis-Pt and enzymatic hydrolysis. Final application of this methodology to commercially available calf thymus DNA samples has been also satisfactorily accomplished.
Introduction
The metabolic pathway of platinum drugs (e.g., cis-Pt, carbo-Pt) in tumoral processes starts with the interaction of such compounds with the DNA nucleobases and the irreversible formation of stable adducts which compromise tumoral cells’ ability to replicate.1,2 In the case of cis-Pt (cis-diamminedichloroplatinum(II)), different mono- and bi-functional covalent adducts can be formed by interaction of this compound with the DNA nucleobases, but the preferred binding sites are N-7 from guanine (G) and adenine (A).3,4 This can be probably ascribed to the high nucleophilicity of the imidazole ring within the purine system. In this regard, the most stable and therefore the most abundant (65%) species is obtained by reaction of one cis-Pt with the two adjacent guanine residues of the same DNA strand.5 The initial step of this process is the hydrolysis of the cis-Pt, which substitutes the first chloride atom by an OH− group that reacts with the nitrogen group of the first guanine. After the monofunctional adduct is formed, the second chloride ligand is hydrolysed and thus the bidentate adduct is generated.6 When the DNA is enzymatically hydrolysed, it should be possible to observe the bidentate complex with the molecular formula [(NH3)2Pt (dGMP)2].Cis-Pt was introduced as a chemotherapeutic drug in the early seventies and the elucidation of its mechanism of action in tumoral cells has been widely studied ever since.7 Although the formation of cis-Pt adducts with the DNA nucleobases has been reported by many authors along the years, ongoing studies on improving the efficiency of Pt-drugs by increasing drug selectivity and minimizing side-effects are still of medical interest.8 Recent studies have revealed a higher number of adducts in the patients responding to Pt drugs and have suggested that the formation of DNA-adducts with cis-Pt could be a pharmacokinetic parameter to optimise in cancer therapy with Pt drugs.9 Therefore, the detection and quantification of DNA-adducts with cis-Pt formed in vivo is of extraordinary present interest.
Considering that the cis-Pt adducts are only formed when the sequence GG and, to a much lesser extent, AG are present, very sensitive techniques are required for the detection of such complexes within DNA samples. In this regard, several tests have been used to evaluate cis-Pt genotoxicity, including those oriented to study changes in DNA integrity such as the COMET assay, TUNEL methods and the number of micronuclei (MNi).10 In addition, the most sensitive method for detecting DNA-adduct formation is based on 32P-post labelling (it is possible to detect 1 adduct in 1010 nucleotides) and it is also applicable to a wide range of DNA lesions.11,12 However, it is a complex assay (including radioactive phosphorus) and experimental conditions can vary depending on the nature of the adduct.
Since the introduction of mass spectrometry (MS), a variety of bioanalytical methods have been used to study the interaction of Pt drugs with DNA, including capillary electrophoresis (CE)13 and liquid chromatography (HPLC) with mass spectrometric (MS) detection.14,15 The increasing use of structural mass spectrometric techniques such as MALDI-MS or electrospray mass spectrometry (ESI-MS) in proteomic studies has been also extended to genomics and, therefore, to DNA interactions with different chemicals.14–16 However, very little work has been published regarding the use of plasma mass spectrometry (ICP-MS) for monitoring DNA interactions in biological samples. This is probably due to the intrinsic limitations associated with phosphorus detection by quadrupole ICP-MS: high ionization potential and so low ionization degree and the existence of polyatomic interferences overlapping the only natural isotope (31P). In this vein, the introduction of high resolution and collision cell ICP-MS technology has allowed the development of several applications revealing the great potential of such ICP-MS systems in detecting phosphorus in P-peptides,17 non-modified nucleotides18,19 and DNA adducts with styrene.20
On the other hand, ICP-MS is probably the most sensitive elemental detector for Pt detection, and this has been used to monitor interactions of Pt drugs with different biomolecules such as proteins.21 Considering the multi-elemental capabilities of ICP-MS, the present work tries to illustrate the advantages of simultaneous P and Pt monitoring, using ICP-MS to detect cis-Pt–DNA adducts after on-line separation from the rest of the nucleotides (only containing P) obtained in DNA hydrolysis. For the optimization of the whole system, the bidentate adduct [(NH3)2Pt(dGMP)2] has been synthesized and the molecular structure of the adducts has been shown by ESI-Q-TOF working in positive and negative modes. The chromatographic separation of cis-Pt-DNA adducts from the unmodified nucleobases has been accomplished by HPLC, with C16 amide with a relatively polar group (–NH–) in the middle of the C16 chain, and a narrow-bore (2 mm id) C8 column. The HPLC-ICP-MS method developed has been satisfactorily applied to the quantitative analysis of the [(NH3)2Pt(dGMP)2] adduct as well as to the free DNA nucleobases in a commercial oligonucleotide after enzymatic hydrolysis. Finally, the presence of the sought cis-Pt adduct in calf thymus DNA, after adequate sample preparation, has been demonstrated by the proposed methodology.
Experimental
Instrumentation
The ICP-MS instrument used in this study was an Agilent 7500 (Agilent Technologies, Tokyo, Japan) equipped with a collision cell system. The instrument was fitted with a PFA micro-nebulizer (Agilent microflow nebulizer) in the case of the C8 column and a conventional Meinhard nebulizer for the C16 column. Instrumental operating conditions are summarized in Table 1.| HPLC parameters | |
| Reverse phase column | C8 5 μm (250 × 2.1 mm id, Alltech Alltima) |
| Mobile phases | 60 mM ammonium acetate (pH = 5.8), 7.5% MeOH |
| Injection volume | 20 μL |
| Flow rate | 0.2 mL min−1 |
| ICP-MS parameters | |
| Forward power | 1500 W |
| External flow | 15 L min−1 |
| Carrier gas flow | 1.1 L min−1 |
| Isotope monitored | 31P,195Pt |
| QP-bias | −11 V |
| Octapole-bias | −13 V |
| Extraction | −3.5 V |
| ESI-MS parameters | |
| Scan type | Negative/positive TOFMS |
| Ionspray voltage | 4.0 kV/5.5 kV |
| Nebulization gas | N2 |
| Injection rate | 5 μL min−1 |
| External calibration | Polypropylene glycol |
| Scan range | m/z 50–1200 |
The HPLC separations were carried out using a dual piston liquid chromatographic pump (Shimadzu LC-10AD, Shimadzu Corporation, Kyoto, Japan) equipped with a sample injection valve, Rheodyne, Model 7125 (Cotati, CA, USA), fitted with a 20 μL injection loop. Two columns were used, a Discovery® RP Amide C16 (150 mm × 4.6 mm, 5 μm) and an Alltech C8 (250 × 2.0 mm id, 5 μm) (Alltima, Alltech Associates, Deerfield, IL, USA).
The ESI-Q-TOF instrument used for this study was a QStar XL model (Applied Biosystems) equipped with the ion-spray source and using N2 as nebulization gas. The scanned range goes from m/z 50–1200: the instrument was calibrated daily and the measurements were taken in negative and positive modes. Final operating conditions are summarized in Table 1.
Reagents and materials
Cis-Pt (cis-diamminedichloroplatinum(II)) and the sodium salts of the four nucleotides (2′-deoxynucleoside 5′-monophosphates of adenosine (dAMP), cytidine (dCMP), guanosine (dGMP) and thymidine (dTMP)) were purchased from Sigma–Aldrich (St. Louis, MO, USA). The mobile phases were prepared by using 18 MΩ cm distilled de-ionized water obtained from a Milli-Q system (Millipore, Bedford, MA, USA). For HPLC separation, ammonium acetate (Riedel-de-Haën, Honeywell Chemicals, Seelze, Germany) salt was dissolved so as to prepare 15 mM and 60 mM solutions, respectively (for C8 and C16 columns) in Milli-Q water. The pH was adjusted by adding acetic acid (Merck, Darmstadt, Germany) dropwise up to a pH of 5.8. Methanol (HPLC grade) was also obtained from Merck. The final separation conditions for both columns are summarized in Table 1.The customized oligonucleotide (231.98 μg) was synthesized by Invitrogen (Invitrogen, Barcelona, Spain) with the sequence 5′-TCCGGTCC-3′ (M.W. = 2362.6 g mol−1) and the Calf Thymus DNA was purchased from Sigma. In both cases, after incubation with cis-Pt, the samples were enzymatically hydrolysed with Nuclease S1 (Amersham Biosciences, Uppsala, Sweden). This enzyme is an endonuclease that catalyses the specific degradation of single-stranded DNA to nucleosides 5′-monophosphate. The activation buffer to dissolve the enzyme was provided by the manufacturer and contained: 10 mM sodium acetate (pH = 4.6), 150 mM sodium chloride, 0.05 mM zinc sulfate and 50% glycerol. To remove the excess of enzyme, membrane ultracentrifugation devices were used (Centricon YM-10 centrifugal filter devices, Millipore).
Procedures
Secondly, another 200 μl of the dissolved oligonucleotide were mixed with 20 μl of cis-Pt solution (75 μg mL−1) and the solutions were vigorously mixed and left to react for 24 hours at 37 °C. After 24 h, 30 μl of the Nuclease S1, previously dissolved on 500 μl of the activation buffer, are added to the reaction mixture and left to react at 45 °C for 14 hours. In the case of DNA, a stock solution was prepared (1 ml from 1 mg of calf thymus DNA in 2 mL of 30 mM acetate buffer, pH = 5.3) and first converted to single-stranded by heating at 97 °C for 30 min. Afterwards, 100 μl of the cis-Pt solution were added to complete the reaction mixture. The solutions were vigorously mixed and left to react for 24 hours at 37 °C, in a similar way to the oligonucleotide mixture, and then the enzyme was added (as previously reported). The excess of enzyme (about 32 kDa) was removed by means of filtration devices (pore size 10![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 Da) and the remaining solution was directly injected in the HPLC system after adequate dilution in the mobile phase.
000 Da) and the remaining solution was directly injected in the HPLC system after adequate dilution in the mobile phase.
269[Gua] − 185212 (r2 = 0.999). Similarly, for quantification of the cis-Pt present in the adduct obtained from the oligonucleotide, a calibration curve was constructed by plotting the theoretical concentration of the synthesized adduct (0, 0.5, 1.75 and 5 μg mL−1 as cis-Pt) versus the peak area, obtained by monitoring 195Pt. A linear adjustment with the equation [Adduct Peak Area] = 200315[cis-Pt] − 56657 (r2 = 0.999).Results and discussion
Synthesis of [(NH3)2Pt(dGMP)2] and electrospray-mass spectrometric characterization
Incubation of the four nucleoside 5′-monophosphates (dAMP, dGMP, dTMP, dCMP) with cis-Pt was carried out independently. As reported by other authors, no reaction was observed in the case of the incubation of cis-Pt with dTMP and dCMP.13 In the case of dAMP, the reaction with cis-Pt provided a very minor signal that could not be completely characterized by ESI-Q-TOF. This can probably be ascribed to the low reaction yield documented for these species (below 30%), rendering any further attempt of identification rather difficult.On the other hand, the incubation of cis-Pt with dGMP provided the ESI-Q-TOF mass spectrum obtained in negative mode that can be seen in Fig. 1 (A and B). Fig. 1(A) shows the full scale mass spectra and here is possible to observe the excess of dGMP without loss of the phosphate group (m/z 346) and its sodium adduct (m/z 368). The dimer of dGMP (m/z 692) and its corresponding sodium adduct (m/z 715) can also be observed. The signal corresponding to the bidentate adduct [(NH3)2Pt(dGMP)2] at m/z 920 can be seen in the magnified spectrum from m/z 900 to m/z 950, although some other clusters show up in the mass spectra, which complicates the clear observation of the Pt isotope pattern. Other important Pt containing fragments are these corresponding to the loss of one ammonia (m/z 904, −17 units with respect to the molecular ion) and two ammonia (m/z 887) groups that can be also observed in the mass spectrum.
![ESI-Q-TOF spectra of the synthesized [(NH3)2Pt (dGMP)2] adduct in negative mode: (A) full scale (magnified m/z 900–950) showing the molecular ion and the sodium adduct; (B) CID spectrum of the ion at m/z 920.](/image/article/2006/JA/b603434a/b603434a-f1.gif) | ||
| Fig. 1 ESI-Q-TOF spectra of the synthesized [(NH3)2Pt (dGMP)2] adduct in negative mode: (A) full scale (magnified m/z 900–950) showing the molecular ion and the sodium adduct; (B) CID spectrum of the ion at m/z 920. | ||
Fig. 1(B) shows the spectra obtained when performing collisional induced dissociation (CID) on the ion of m/z 921. The most abundant product ions arise from the loss of one and two ammonia groups from the molecular ions at m/z 904 and 887, respectively. Other platinated product ions were of very weak relative abundances since they are either neutral (due to the loss of a phosphate group and the two positive charges on the platinum) or positively charged (loss of the two phosphate groups). Therefore, they show low intensity in negative mode although they can be partially observed at m/z 540 and 495. The highest signal at m/z 346 is due to the guanosine 5′-monophosphate.
For a more complete understanding of the fragmentation pathway of the synthesized adduct, the same experiment described above was conducted in positive mode. The obtained results are shown now in Fig. 2 (Fig. 2(A) corresponds to the MS and Fig. 2(B) to the CID spectrum on the ion at m/z 922). Fig. 2(A) exhibits the Pt isotope pattern at m/z 922 (calculated mass resolution of 10300) as well as at m/z 944 corresponding to the sodium adduct of the [(NH3)2Pt(dGMP)2]. In the case of the CID spectrum (shown in Fig. 2(B)) different platinated product ions can be observed, mainly obtained from successive fragmentations of the ions at m/z 905 and 888 that correspond to the loss of one and two ammonia groups, respectively. For clarity, the identified fragment ions are listed in Table 2. From the [(NH3)1Pt(dGMP)2]+ ion at m/z 905, several fragments can be obtained at m/z 380, 460, 513, 540, 691, 709 and 790 with the structures detailed in Table 2 and marked with bold figures on the mass spectrum. Similarly, the main ions arising from the fragmentation of [Pt(dGMP)2]+ at m/z 888 are also listed in Table 2 with the corresponding structures at m/z 443, 496, 523, 575, 674 and 772 and marked as shaded figures on the mass spectrum. Very few fragments could be identified as product ions of the molecular ion (m/z 922 and m/z 557, see Table 2) which seem to be labile under these conditions, releasing one or two ammonia groups. In any case, the results obtained in both positive and negative modes indicate the presence of the expected bidentate complex of guanine with cis-Pt.
![ESI-Q-TOF spectra of the synthesized [(NH3)2Pt(dGMP)2] adduct in positive mode: (A) full scale (magnified m/z 900–950), (B) CID spectrum of the ion at m/z 922. Bold figures arising from the fragmentation of [(NH3)1Pt(dGMP)2]+ and shaded figures coming from the fragmentation of [Pt(dGMP)2]+.](/image/article/2006/JA/b603434a/b603434a-f2.gif) | ||
| Fig. 2 ESI-Q-TOF spectra of the synthesized [(NH3)2Pt(dGMP)2] adduct in positive mode: (A) full scale (magnified m/z 900–950), (B) CID spectrum of the ion at m/z 922. Bold figures arising from the fragmentation of [(NH3)1Pt(dGMP)2]+ and shaded figures coming from the fragmentation of [Pt(dGMP)2]+. | ||
| Precursor ion | |||
|---|---|---|---|
| Fragments | [Pt(dGMP)2(NH3)2]+ | [Pt(dGMP)2(NH3)]+ | [Pt(dGMP)2]+ |
| a Gua = Guanine, purine base, P = phosphate group, r = ribose. | |||
| Gua + H2O | — | 380 | — |
| rGua | — | 460 | 443 |
| (Gua) (Gua) | — | 513 | 496 |
| prGua or GMP | 557 | 540 | 523 |
| (prGua) (Gua) | — | 691 | 674 |
| (prGua) (Gua) + H2O | — | 709 | 692 |
| (prGua) (rGua) | — | 789 | 772 |
| (prGua) (prGua) | 922 | 905 | 888 |
| (prGua) (prGua) − H2O | — | 870 | |
Chromatographic separation nucleoside 5′-monophosphates and the cis-Pt adduct
Once the synthesized cis-Pt adduct had been positively identified, the next step was to develop a hyphenated methodology in order to be able to separate the adduct from the free nucleobases, which would be present at high levels in DNA samples. The coupling of liquid chromatography to ICP-MS as an element selective detector (for 31P and 195Pt) creates the ideal combination since it provides the sensitivity and specificity necessary for this type of application. For this purpose, two different chromatographic columns have been evaluated:A similar effect was observed when the percentage of methanol was increased from 2.5 to 10% (shorter retention times at higher percentages of methanol). The variation of the ionic strength (from 15 to 150 mM) caused very minor changes to the retention time/peak shape of the adduct and the nucleobases. Therefore, 15 mM ammonium acetate, pH = 5.8 and 5% MeOH (flow 1 ml min−1) were selected as optimum conditions for the separation of the complex from the nucleobases and Fig. 3 shows the chromatogram obtained by monitoring 31P and 195Pt signals on the ICP-MS. Under these conditions, no good resolution was obtained among nucleobases (only containing 31P) eluting from 2 to 6 min, although all of them were well separated from the [(NH3)2Pt(dGMP)2] adduct (tr = 12.64 min, P and Pt are co-eluting). Unfortunately, the presence of the amide groups on the stationary phase of this column seem to have a strong effect on the retention of the adduct that exhibited a tailing profile under any set of working conditions (see Fig. 3, with a peak width at half height of about 1 min).
![HPLC-ICP-MS chromatogram of the separation of DNA-nucleobases and the synthesized adduct [(NH3)2Pt (dGMP)2] using the Discovery amide C16 column showing the 31P and 195Pt traces. The inset shows the structure of the synthesized adduct. Chromatographic conditions summarized in Table 1.](/image/article/2006/JA/b603434a/b603434a-f3.gif) | ||
| Fig. 3 HPLC-ICP-MS chromatogram of the separation of DNA-nucleobases and the synthesized adduct [(NH3)2Pt (dGMP)2] using the Discovery amide C16 column showing the 31P and 195Pt traces. The inset shows the structure of the synthesized adduct. Chromatographic conditions summarized in Table 1. | ||
![HPLC-ICP-MS chromatograms corresponding to the separation of (A) traces of 31P and 195Pt from the [(NH3)2Pt(dGMP)2] adduct and (B) the four nucleosides 5′-monophosphates using the narrow-bore C8 column.](/image/article/2006/JA/b603434a/b603434a-f4.gif) | ||
| Fig. 4 HPLC-ICP-MS chromatograms corresponding to the separation of (A) traces of 31P and 195Pt from the [(NH3)2Pt(dGMP)2] adduct and (B) the four nucleosides 5′-monophosphates using the narrow-bore C8 column. | ||
Quantitative analysis of cis-Pt adducts in oligonucleotides and DNA samples
In order to study the suitability of the proposed hybrid methodology to the on-line analysis of cis-Pt adducts in DNA samples, a first experiment was conducted using a customized oligonucleotide, as model, with a known base sequence (TCCGGTCC). This structure contains two adjacent guanine units within the same strand and should induce the formation of the [(NH3)2Pt(dGMP)2] adduct (once it had been treated with cis-Pt and enzymatically hydrolyzed). In order to test the dGMP concentration provided by the manufacturer and also the efficiency of the enzymatic hydrolysis, an aliquot of the oligonucleotide was treated with Nuclease S1. This enzyme cleaves single stranded DNA nucleobases, releasing nulceosides 5′-monophosphate. After removing the excess of enzyme, the sample was analysed by RPLC-ICP-MS (with the narrow-bore C8) and monitoring of 31P signals in the ICP-MS. Fig. 5 shows the chromatogram corresponding to the individual nucleobases cleaved from the oligonucleotide and analysed by RPLC-ICP-MS, monitoring 31P. As can be seen, chromatographic peaks matching the retention times of dCMP, dTMP and dGMP, as well as inorganic phosphate, have been obtained. Quantitative analysis of the dGMP present in the oligonucleotide was performed by linear calibration using different concentrations of the dGMP standard and turned out to be 13 ± 1 μg mL−1. According to the manufacturer’s specifications the dGMP content was around 9.4 μg mL−1 although no uncertainty was provided. Thus, our obtained value, although slightly higher, can be considered acceptable and proves the suitability of the used enzyme.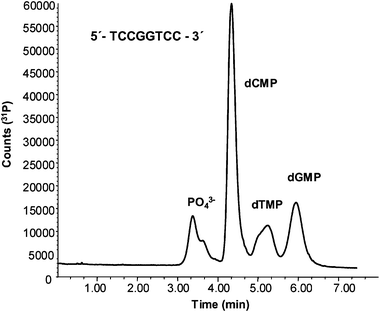 | ||
| Fig. 5 Separation of the nucleosides 5′-monophosphate obtained from the enzymatic hydrolysis of the oligonucleotide (5′-TCCGGTCC-3′) by monitoring 31P. Chromatographic conditions summarized in Table 1. | ||
Additionally, incubation of the oligonucleotide with cis-Pt was carried out as described in the procedures section. Then, the mixture was enzymatically hydrolysed and analysed by HPLC-ICP-MS. The results obtained can be observed in Fig. 6(A). In this case, together with the signals corresponding to dCMP, dTMP, dGMP (unreacted) and inorganic phosphate, it is possible to observe the presence of a P–Pt containing species at 26 min. The shift in the retention time in respect of the synthesized adduct (tr, 18 min) can be ascribed to the limitations of the enzyme when cleaving the internal phosphate group between the adjacent guanines.22 This implies that the structure of the obtained adduct is slightly different from the previously synthesized compound as proved by ESI-Q-TOF (see inset of Fig. 6(A)). In this case, the molecular ion [M + H]+ at m/z 904 and the sodium adduct [M + Na]+ at m/z 926, exhibiting the Pt isotope pattern, correspond to the proposed structure with the internal phosphate bridge between guanines (inset Fig. 6(B)). In this case, no signal was obtained at m/z 922 (molecular ion of the synthesized adduct, see Fig 2(A)). In any case, the signal on the ICP-MS for P and Pt should be independent of the structure, and therefore by performing a calibration curve using different concentrations of the synthesized [(NH3)2Pt(dGMP)2] it is possible to estimate the concentration of the cis-Pt in the obtained adduct. This concentration turned out to be 1.65 ± 0.4 μg mL−1, which is in good agreement with the incubated cis-Pt (2 μg mL−1) if the slight amount of free cis-Pt eluting at about 6.5 min (see Fig. 6(A)) is taken into account.
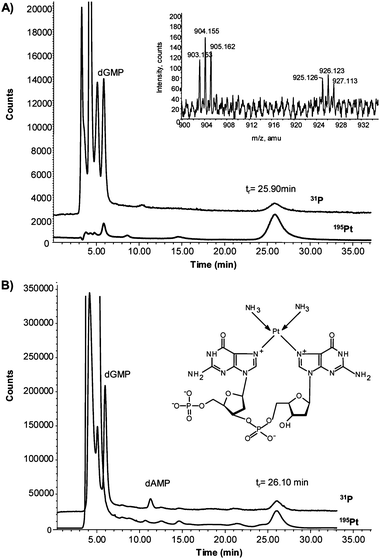 | ||
| Fig. 6 Chromatogram obtained by HPLC-ICP-MS (narrow-bore C8 column) from the incubation with cis-Pt of (A) oligonucleotide (5′-TCCGGTCC-3′) and (B) calf thymus DNA, both after enzymatic hydrolysis with Nuclease S1. The inset of Fig. 6(A) shows the proposed structure of the Pt–P complex found in both samples. | ||
Considering the stoichiometry of the complex (two dGMP units for each cis-Pt), it is also possible to calculate the dGMP concentration in the obtained adduct, which was estimated to be 3.3 μg mL−1. Using the previous dGMP calibration curve, it is also possible to calculate the unreacted dGMP in the oligonucleotide (elutes at 6 min) obtaining a concentration of 6.9 μg mL−1. When the free and the reacted guanine are added together, the final concentration turned out to be about 10.2 ± 0.5 μg mL−1, a figure also in good agreement with the recommended concentration given by the manufacturer (9.4 μg mL−1).
Similarly, DNA studies with commercially available calf thymus DNA were conducted. This is double-stranded DNA with unlabelled sequence. Therefore, as a first step, double-stranded was converted to single-stranded DNA by being heated at 95 °C and immediately afterwards incubated with cis-Pt. Then, the sample was processed as described above for the oligonucleotide. The chromatographic profile obtained for P and Pt in the DNA sample is shown in Fig. 6(B). As can be seen, the main Pt–P containing species elutes again at about 26 min (that is, shifted with respect to the previously synthesized adduct). As in the case of the adduct formed with the oligonucleotide (they have mainly the same retention time), this shift can be ascribed to a remaining phosphate group bridging the two guanines in the adduct obtained from the DNA (see structure in the inset of Fig. 6(B)). This possibility has also been shown by other authors5,22 using similar enzymes and it could be related to the stereochemical configuration of the formed adduct.
Conclusions
The synthesis and structural characterization of the complex formed between pure dGMP and cis-Pt, (NH3)2Pt(dGMP)2, has been successfully accomplished by ESI-Q-TOF. Although, the molecular ion was detected in both positive and negative modes, more complete information about the fragmentation pathway of the molecule has been obtained by working in the positive mode. In this case, a rich spectrum with ions arising from the fragmentation of the molecular ion after the subtraction of one and two ammonia groups has been obtained. Concerning the separation of the synthesized (NH3)2Pt(dGMP)2 from the DNA nucleobases, good results have been accomplished by using a narrow bore C8 column and a mobile phase containing 60 mM ammonium acetate (pH = 5.8) and 7.5% MeOH. By performing the on-line coupling of this column with ICP-MS through a micro-nebulizer, it is possible to observe the simultaneous 31P and 195Pt signals and, therefore, to characterize the retention time of the formed adduct and the different nucleobases.Finally, the developed HPLC-ICP-MS methodology has been applied to the quantification of DNA adducts in a custom oligonucleotide and in calf thymus DNA. In the case of the oligonucleotide, acceptable results have obtained in comparison with the recommended manufacturer value. Therefore, estimations of the concentration of the cis-Pt adduct in real-life DNA samples is possible. However, it appears that the limitations of the enzymatic hydrolysis to cleave the internal phosphodiester bridge between the two adjacent guanines (once they are forming the complex with cis-Pt), proved by ESI-Q-TOF in the oligonucleotide, justify the observed shifting in the retention time of the adduct (with respect to the synthesized one).
Acknowledgements
The authors want to thank the financial support for this work through the project CTQ2004–03005 from the Ministry of Education and Science and the Regional Government for D.G.S.References
- S. E. Sherman and S. J. Lippard, Chem. Rev., 1987, 87, 1153–1181 CrossRef CAS.
- S. Aebi, B. Kurdi-Haidar, R. Gordon, B. Cenni, H. Zheng, D. Howell, R. D. Christen, C. R. Boland, M. Koi, R. Fishel and S. B. Howell, Cancer Res., 1996, 56, 3087–3090 CAS.
- A. Zenker, M. Galanski, T. L. Bereuter, B. K. Keppler and W. Lindner, J. Chromatogr. A, 1999, 852, 337–346 CrossRef CAS.
- T. Hagemeister and M. Linscheid, J. Mass Spectrom., 2001, 37, 731–747.
- H. Iijima, H. B. Patrzyc, J. B. Dawidzik, E. E. Budzinski, H.-C. Cheng, H. G. Freund and H. C. Box, Anal. Biochem., 2004, 333, 65–71 CrossRef CAS.
- Principles of Bioinorganic Chemistry, eds. S. J. Lippard and J. Berg, University Science Books, CA, USA, 1994 Search PubMed.
- E. Reed, Cisplatin, Cancer Chemother. Biol. Response Modifiers, 1999, 18, 144–151 Search PubMed.
- H. J. Wanebo and J. F. Belliveau, Cancer Chemother. Pharmacol., 1999, 43, 427–434 CrossRef CAS.
- E. Reeds, R. F. Ozols, R. Tarone, S. H. Yuspa and M. C. Poirier, Proc. Natl Acad. Sci. USA, 1987, 84, 5024–5028 CrossRef.
- K. Jirsova, V. Mandys, W. H. Gispen and P. R. Bar, Neurosci. Lett., 2006, 392, 22–26 CrossRef CAS.
- D. H. Phillips and M. Castegnaro, Mutagenesis, 1999, 14, 301–315 CrossRef CAS.
- M. Zeisig and L. Möller, J. Chromatogr., B: Biomed. Appl., 1997, 691, 341–350 CrossRef CAS.
- U. Warnke, J. Gysler, B. Hofte, U. R. Tjaden, J. van der Greef, C. Kloft, W. Schunack and U. Jaehde, Electrophoresis, 2001, 22, 97–103 CrossRef CAS.
- V. Beljanski, J. M. Villanueva, P. W. Doetsch, G. Natile and L. G. Marzilli, J. Am. Chem. Soc., 2005, 127, 15833–15842 CrossRef CAS.
- A. C. Gingras, M. Caballero, M. Zarske, A. Sanchez, T. R. Hazbun, S. Fields, N. Sonenberg, E. Hafen, B. Raught and R. Aebersold, Mol. Cell. Proteomics, 2005, 4, 1725–1740 Search PubMed.
- P. Iannitti-Tito, A. Weimann, G. Wickham and M. M. Sheil, Analyst, 2000, 125, 627–634 RSC.
- D. Pröfrock, P. Leonhard, W. Ruck and A. Prange, Anal. Bioanal. Chem., 2005, 381, 194–204 CrossRef CAS.
- C. Siethoff, I. Feldmann, N. Jakubowski and M. Linscheid, J. Mass Spectrom., 1999, 31, 421–426 CrossRef.
- D. Pröfrock, P. Leonhard and A. Prange, J. Anal. At. Spectrom., 2003, 18, 708–713 RSC.
- M. Edler, N. Jakubowski and M. Linscheid, Anal. Bioanal. Chem., 2005, 381, 205–211 CrossRef CAS.
- G. S. Yang, R. Miao, C. Jin, Y. H. Mei, H. W. Tang, J. Hong, Z. J. Guo and L. G. Zhu, J. Mass Spectrom., 2005, 40, 1005–1016 CrossRef CAS.
- R. Gupta, J. L. Beck, M. M. Sheil and S. F. Ralph, J. Inorg. Biochem., 2005, 99, 552–559 CrossRef CAS.
Footnote |
| † Presented at the 2006 Winter Conference on Plasma Spectrochemistry, Tucson, AZ, USA, January 8–14, 2006. |
| This journal is © The Royal Society of Chemistry 2006 |