Precise ruthenium fission product isotopic analysis using dynamic reaction cell inductively coupled plasma mass spectrometry (DRC-ICP-MS)†
Christopher F. Brown*, P. Evan Dresel, Keith N. Geiszler and Orville T. Farmer
Pacific Northwest National Laboratory, P.O. Box 999, MSIN P7-22, Richland, Washington 99352, USA. E-mail: christopher.brown@pnl.gov; Fax: (509) 376-4890; Tel: (509) 376-8389
First published on 9th May 2006
Abstract
99Tc is a contaminant of interest at numerous nuclear facilities because it is quite mobile in sub-surface environments and is a key contributor to long-term risk. However, as a mono-isotopic fission product, 99Tc is limited in its use as a signature to differentiate between different waste disposal pathways that could have contributed to sub-surface contamination at these facilities. Ruthenium fission-product isotopes are attractive analogues for the characterization of 99Tc sources because of their direct similarity to technetium with regard to subsurface mobility, their large fission yields, and low natural background concentrations. We developed an inductively coupled plasma mass spectrometry (ICP-MS) method capable of measuring ruthenium isotopes in groundwater samples and water extracts of vadose zone sediments. Samples were analyzed directly on a PerkinElmer ELAN DRC II ICP-MS after a single pass through a 1-ml bed volume of Dowex AG 50W-X8 100–200 mesh hydronium-based cation exchange resin. Precise ruthenium isotopic ratio measurements were achieved using a low-flow Meinhard-type nebulizer and long sample acquisition times (150![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 ms). Relative standard deviations of triplicate replicates were maintained at less than 0.5% when the total ruthenium solution concentration was 0.1 ng ml−1 or higher. Further work was performed to minimize the impact caused by mass interferences using the dynamic reaction cell (DRC) with O2 as the reaction gas. Aqueous concentrations of 96Mo and 96Zr, two potential interferents to the analysis of total ruthenium concentration, were reduced by more than 99.7% in the reaction cell prior to injection of the sample into the mass analyzer quadrupole. The DRC was used in combination with mass correction to quantitatively analyze samples containing up to two orders of magnitude more zirconium and molybdenum than ruthenium. The analytical approach documented herein provides an efficient and cost-effective way to precisely measure ruthenium isotopes and quantitate total ruthenium (natural versus fission-product) in aqueous matrixes.
000 ms). Relative standard deviations of triplicate replicates were maintained at less than 0.5% when the total ruthenium solution concentration was 0.1 ng ml−1 or higher. Further work was performed to minimize the impact caused by mass interferences using the dynamic reaction cell (DRC) with O2 as the reaction gas. Aqueous concentrations of 96Mo and 96Zr, two potential interferents to the analysis of total ruthenium concentration, were reduced by more than 99.7% in the reaction cell prior to injection of the sample into the mass analyzer quadrupole. The DRC was used in combination with mass correction to quantitatively analyze samples containing up to two orders of magnitude more zirconium and molybdenum than ruthenium. The analytical approach documented herein provides an efficient and cost-effective way to precisely measure ruthenium isotopes and quantitate total ruthenium (natural versus fission-product) in aqueous matrixes.
Introduction
99Tc has been recognized as a contaminant of concern at numerous federal, commercial, and international facilities1–6 due to its long half-life (213000 years) and high mobility in sub-surface environments in its predominant anionic form, the pertechnetate ion (TcO4−).1,7–12 At the U.S. Department of Energy’s Hanford Site, located in southeastern Washington State, 99Tc was produced as a by-product of nuclear fission in the site’s nine plutonium production reactors. Both inadvertent and planned releases of waste solutions to the environment have resulted in widespread 99Tc contamination in the Hanford groundwater system.13 Recently, 99Tc was observed in groundwater samples collected from a monitoring well near the T single-shell tank farm‡ at concentrations in excess of 6660 Bq L−1 (the current drinking water standard is 33.3 Bq L−113). Discovery of these levels of 99Tc in groundwater near the T single-shell tank farm has prompted studies focused on identifying the source of the 99Tc. However, as a mono-isotopic fission product,1499Tc lacks the inherent ability to be used as a signature to differentiate between different waste disposal pathways (i.e., liquid waste discharges to cribs, ponds, and trenches versus leaks from waste storage tanks).Several other fission-product isotopes are similar in mobility to 99Tc and may help in identifying the 99Tc sources. These include fission-product isotopes of selenium, molybdenum, ruthenium, rhodium, and palladium. However, the stable ruthenium fission-product isotopes (101Ru, 102Ru, and 104Ru) are principally attractive because of their direct similarity to conservative species, such as 99Tc, with regard to leaching/mobility,15 their large fission yields,16 and their low natural background concentrations.17,18 Additionally, ruthenium fission isotopes are particularly sensitive to variation in reactor conditions and fuel type and, therefore, may help constrain possible contaminant sources16 (Fig. 1).
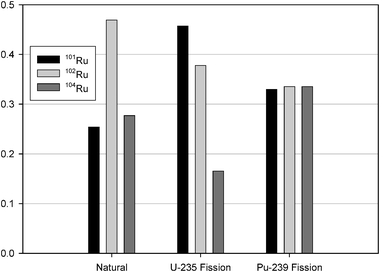 | ||
| Fig. 1 Variation in ruthenium isotopic abundances for natural, 235U fission-produced, and 239Pu fission-produced 101Ru, 102Ru, and 104Ru. 235U and 239Pu fission-produced isotopic ratios are dependent upon reactor power and exposure time. | ||
High-precision ruthenium isotopic analyses typically require either sophisticated analytical equipment that is not available to the general scientific community,19,20 or labor-intensive separation and pre-concentration steps prior to sample analysis.21,22 However, technological advancements in the field of quadrupole-based inductively coupled plasma mass spectrometry (ICP-MS) have enabled this type of instrument to become an important resource in the analysis of ruthenium and ruthenium isotopic ratios.17,23 PerkinElmer’s ELAN DRC II (PerkinElmer Sciex, Shelton, CT) is a state-of-the-art ICP-MS that uses Dynamic Reaction Cell (DRC) technology to eliminate polyatomic and mass interferences.24–26 Specifically, DRC technology could prove particularly useful for the analysis of total ruthenium, which requires the removal of other mass interferences, such as 96Mo, 98Mo, and 96Zr from natural ruthenium, as well as 100Mo, 102Pd and 104Pd from natural and fission-produced ruthenium.
Molybdenum was reported as a trace constituent (present at concentrations of less than 4 mg kg−1 of dry sediment) in uncontaminated vadose zone sediment collected in the vicinity of the T tank farm.18 Groundwater samples collected from the Hanford Site during fiscal year 2005 contained dissolved molybdenum at concentrations ranging from 28.6 to 203 ng ml−1.13 Similarly, zirconium was reported as a minor constituent (present at concentrations of less than 0.037 wt% as ZrO2) in uncontaminated vadose zone sediment collected in the vicinity of the T tank farm.18 Groundwater samples collected from the Hanford Site during fiscal year 2005 were not monitored for dissolved zirconium; however, it was assumed that zirconium was not present at detectable concentrations in the samples given the low solubility of ZrO2 under environmental pH conditions. Finally, palladium was not detected in vadose zone sediment collected in the vicinity of the T tank farm,18 so any contribution of palladium at atomic mass units 102 and 104 was considered negligible. Further, since the analysis of ruthenium at atomic mass units 98 and 100 can be affected by fission produced 98Mo and 100Mo, 96Ru is the preferred mass for quantifying the concentration of natural ruthenium in samples containing fission product isotopes. Therefore, 96Mo and potentially 96Zr are the primary mass interferences that must be removed in order to make quantitative analysis of total ruthenium possible.
We developed an ICP-MS analytical method capable of precisely measuring ruthenium isotopes in groundwater samples and extracts of vadose zone sediments. The isotopes 101Ru, 102Ru, and 104Ru are the only fission isotopes of significance since the production of lower mass isotopes is largely blocked by stable molybdenum and 99Tc; 103Ru and 106Ru are not of concern in aged wastes because of their short (39 day and 368 day) half-lives, respectively. Samples were analyzed directly on a PerkinElmer ELAN DRC II ICP-MS after a single pass through a 1-ml bed volume of Dowex AG 50W-X8 100-200 mesh hydronium-based cation exchange resin. Further work was performed to minimize the potential interferences caused by molybdenum and zirconium using the DRC with O2 as the reaction gas. This paper describes our success at minimizing 96Mo and 96Zr as mass interferences, as well as the accuracy and precision of the analytical technique. The analytical approach documented herein provides an efficient and cost-effective way to precisely measure ruthenium isotopes and quantitate total ruthenium (natural versus fission-product derived) in aqueous matrixes.
Experimental
Chemicals
All chemicals and solutions were prepared using distilled water further treated using a Millipore system (Billerica, MA) with a minimum resistivity of 18 MΩ-cm. Optima grade nitric acid (Fisher Scientific, Pittsburgh, PA) was used without additional purification. Dowex AG 50W-X8 100–200 mesh cation exchange resin (Fisher Scientific) was cleaned with three bed volumes of 16 M Optima nitric acid and ten bed volumes of 18 MΩ-cm de-ionized water prior to use. A single-element ruthenium (Ru) standard purchased from Ultra Scientific, Inc. (North Kingstown, RI) was used for initial method development, and to calibrate the ICP-MS. Once calibrated, continuing calibration verification checks were performed using a single-element Ru standard purchased from CPI International (Santa Rosa, CA). Single-element molybdenum (Mo) and zirconium (Zr) standards, both purchased from Ultra Scientific, Inc., were used for DRC method development. Indium (In), which was used as the internal standard during all analytical runs, was also purchased as a single element standard from Ultra Scientific, Inc. Research grade oxygen (99.999% pure), purchased from Norco Specialty Gases (Boise, ID), was used during testing and analysis in the DRC mode.Equipment used
Groundwater samples were filtered in the field using 0.45 μM filters and were stored unpreserved at 4 °C in high density polyethylene (HDPE) or perfluoralkoxy copolymer (PFA) bottles. Vadose zone sediment samples were collected in 10-cm diameter Lexan liners and were stored at 4 °C until the time of processing. Water extraction of the sediment was performed in 250-ml HDPE or PFA bottles, and all extracts were filtered using 0.45 μM filters and preserved with 1% by volume Optima nitric acid. Bio-Rad Poly-Prep® chromatography columns packed with Dowex resin were used to perform cation separation of all aqueous samples prior to analysis by ICP-MS.Cleaning of labware
All labware was cleaned prior to use to reduce interferences and to minimize cross-contamination. The labware was first rinsed in 18 MΩ-cm de-ionized water to remove dust and loose particles. This was followed by soaking the labware in 0.1% by volume ammonium hydroxide for 1–2 hours. After soaking, the labware was rinsed in de-ionized water and soaked in a 5% by volume Optima nitric acid solution for 1–2 hours. The labware was again rinsed with de-ionized water and allowed to air dry under protective covering prior to use.Samples
Samples analyzed as part of this study included groundwater and vadose zone sediment collected from the Hanford Site (Fig. 2). Approximately half of the groundwater samples were collected in March of 2005 from a network of shallow monitoring wells (screened at the top of the aquifer) located around the T tank farm.13 The remaining groundwater samples were depth-discrete profile samples collected as deep as 150 feet below the water table in well 299-W11-25B, which was installed near the T tank farm during fiscal year 2005.27 The vadose zone sediments analyzed as part of this study were depth-discrete samples collected from two bore holes (C4104 and C4105) emplaced within the tank farm, near a documented leak from Tank T-106, during fiscal year 2003.18 | ||
| Fig. 2 Map showing sampling locations; the dashed line represents the fence line boundary of the T tank farm. | ||
Sample preparation
The vadose zone sediments were processed using a 1∶1 sediment∶de-ionized water extraction method adapted from Thomas.28 The 1∶1 sediment∶de-ionized-water extracts were prepared by adding an exact weight of de-ionized water to approximately 60–80 g of field-moist sediment. The weight of de-ionized water needed was calculated based on the weight of the field-moist samples and their previously determined moisture contents. The sum of the existing pore water and the de-ionized water was fixed at the mass of the oven-dry sediment. An appropriate amount of de-ionized water was added to HDPE or PFA screw cap jars containing the sediment samples. The jars were sealed and briefly shaken by hand, then placed on a mechanical orbital shaker for one hour. The samples were allowed to settle until the supernatant liquid was fairly clear. The supernatant was carefully decanted and filtered through 0.45 μm membranes.Aliquots of several of the vadose zone sediments were packed into drainable cells that were inserted into an unsaturated flow apparatus (UFA) ultracentrifuge. The samples were centrifuged for up to 8 hours at several thousand g’s to extract the pore water out of the sediment. The pore waters were filtered through 0.45 μm membranes and analyzed for ruthenium isotopes for comparison against the 1∶1 sediment∶de-ionized water extracts.
The groundwater samples, sediment:de-ionized water extracts, and UFA samples were processed through columns containing the Dowex cation exchange resin. Sufficient sample volume was pretreated with the cation resin to generate 10 ng of total ruthenium in the aliquot that was analyzed. For low concentration samples, 10 ml of sample solution was used, which resulted in the final solution containing less than 10 ng of total ruthenium.
Instrumentation and analysis of ruthenium
Direct analysis of the treated samples was performed via inductively coupled plasma mass spectrometry. Calibration standards were prepared by diluting the 1000 mg L−1 ruthenium certified stock standard into appropriate volumes of 2% Optima nitric acid solution containing 1 ng ml−1 indium as the internal standard; internal standards are chosen based on their proximity (atomic mass) to an element of interest, and are used to verify instrument performance and correct for instrument drift. An independent calibration check standard was prepared from the 1000 mg L−1 ruthenium independent certified stock standard in 2% Optima nitric acid. Two percent Optima nitric acid was used to prepare instrument blanks, and was also used as the rinse solution between each sample injection throughout the sample analysis.All solution samples were analyzed using a PerkinElmer ELAN DRC II ICP-MS equipped with a dual-channel gas manifold. Plasma power was set to 1000 W, with a plasma gas flow of 15 ml min−1 and an auxiliary gas flow of 1.05 ml min−1. Either a standard Meinhard-type nebulizer with a gas flow of 0.98 ml min−1 or a low-flow Meinhard-type nebulizer with a gas flow of 0.9 ml min−1 was used for sample introduction. The PerkinElmer ELAN DRC II comes with a quartz cyclonic spray chamber as standard. Sample introduction was maintained at 1.0 ml min−1 for the standard nebulizer via an integrated peristaltic pump and at 0.25 ml min−1via self-aspiration for the low-flow nebulizer. Oxygen reaction gas, when used, was added through channel B of the gas manifold at a flow rate of 0.8 ml min−1. The rejection parameter q (rpq), which adjusts the RF voltage applied to the reaction cell quadrupole, was set to 0.55 for molybdenum and 0.4 for ruthenium and zirconium.
Results and discussion
Instrument optimization
A solution containing 5 ng ml−1 total ruthenium in 2% nitric acid was analyzed in standard mode (i.e., no reaction gas was used) with peak hopping data acquisition using a 1-ml Meinhard-type nebulizer, and the percent relative standard deviation data for three-replicate samples are presented in Table 1. The three default isotopes for 101Ru (100.9), 102Ru (101.9), and 104Ru (103.9) programmed into the ELAN software were the only atomic mass units investigated during the initial instrument optimization work. When the instrument was operated under high-throughput acquisition parameters, with a dwell time of 50 ms and 10 sweeps, the precision of the replicate analyses was unsatisfactory. The relative standard deviations for all three of the isotopes monitored were in excess of 3%. However, with only minor adjustments to the dwell time and sweeps, the statistical precision of analysis was markedly improved. Increasing the dwell time by a factor of 10–500 ms and decreasing the sweeps to 5 for a total acquisition time of 7500 ms per sample (2500 ms × 3 replicates) resulted in relative standard deviations ranging from 0.253 to 0.402% for the three isotopes. While all of the relative standard deviations were below the target level of 0.5%, measuring samples containing 5 ng ml−1 total ruthenium would require significant sample pre-concentration for the present study and was not realistic for the vadose zone or groundwater samples where limited sample was available.| Solution concentration/ng ml−1 | 101Ru relative standard deviation (%)c | 102Ru relative standard deviation (%) | 104Ru relative standard deviation (%) |
|---|---|---|---|
| a Data was acquired with a dwell time of 50 ms and 10 sweeps.b Data was acquired with a dwell time of 500 ms and 5 sweeps.c Standard deviations are based upon n = 3 measurements. | |||
| 5.0a | 3.13 | 3.33 | 3.34 |
| 5.0b | 0.402 | 0.272 | 0.253 |
To enhance precision while analyzing low concentration samples, the PerkinElmer ELAN DRC II instrument was retrofitted with a low-flow, self-aspirating, Meinhard-type nebulizer. Use of a low-flow nebulizer, with a flow-rate of 0.25 ml min−1, enabled the total acquisition time for each sample to be increased significantly (150000 ms). The percent relative standard deviation results for three replicate analyses of a 0.1 ng ml−1 total ruthenium solution are presented in Table 2. Contained in the table are two sets of data for the 0.1 ng ml−1 ruthenium standard; the first set of data was acquired after a standard instrument warm-up time (i.e., 2 hours), while the second set was acquired after the instrument was allowed to warm-up for 24 hours. Also contained in Table 2 are results for additional atomic mass units of ruthenium (101Ru, 102Ru, and 104Ru), which were measured to investigate instrument stability off-peak from the default atomic mass units programmed into the ELAN software: 100.9Ru, 101.9Ru, and 103.9Ru. After the standard instrument warm-up time, the precision of replicate isotopic measurements ranged from a low of 0.262% to a high of 1.07%. It should be noted that the most stable location to measure 104Ru after standard warm-up time was slightly off-peak from the default mass of 103.9, indicating that the atomic mass unit providing the highest intensity signal was not always the most stable location to perform the analysis.
| Solution concentration/ng ml−1 | 100.9Rud RSDc (%) | 101Ru RSD (%) | 101.9Rud RSD (%) | 102Ru RSD (%) | 103.9Rud RSD (%) | 104Ru RSD (%) |
|---|---|---|---|---|---|---|
| a Data was acquired with a dwell time of 1000 ms, 50 sweeps, and 2 hours of instrument warm-up time.b Data was acquired with a dwell time of 1000 ms, 50 sweeps, and 24 hours of instrument warm-up time.c RSD calculations are based upon n = 3 measurements.d Indicates default instrument atomic mass unit. | ||||||
| 0.1a | 0.262 | 0.720 | 0.348 | 0.428 | 1.07 | 0.393 |
| 0.1b | 0.197 | 0.446 | 0.144 | 0.375 | 0.167 | 0.168 |
When the instrument was allowed to warm-up for an extended period of time (24 hours), the precision of analysis increased significantly for all of the atomic mass units analyzed. Under this scenario, the relative standard deviations ranged from 0.144 to 0.446%, with the ELAN default ruthenium isotopes having relative standard deviations ranging from 0.144 to 0.197%. With the extended warm-up time, the peak maximum location proved to be the most stable location to analyze each ruthenium isotope. Finally, the measured precision of a 0.1 ng ml−1 total ruthenium solution, at less than 0.2% relative standard deviation, using the extended acquisition time of 150000 ms, was a factor of three times better than previously reported results using quadrupole-based ICP-MS,17 and would enable the analysis of all but the most dilute groundwater samples to be performed without the requirement of sample pre-concentration.
Sample analysis
Table 3 and Fig. 3 contain the 101Ru∶104Ru and 102Ru∶104Ru ratio data for the groundwater samples and vadose zone sediment extracts (water extracts and UFA samples) based on background subtraction and includes an alpha correction factor. The alpha correction was calculated from the natural ruthenium standards by dividing the measured relative abundance of each isotope of interest (101Ru, 102Ru, and 104Ru) by the literature value for the isotope’s relative abundance in natural ruthenium. The relative abundance is the amount of each isotope (101Ru, 102Ru, and 104Ru) divided by the sum of concentration of the three isotopes measured. Thus, the amounts of other ruthenium isotopes, which are subject to isobaric interferences and are not easily measured, do not affect the abundance calculation of the three isotopes that were measured. The measured count per second data from isotopes 101Ru, 102Ru, and 104Ru in the samples were then corrected by dividing them by the alpha correction factor for each respective isotope prior to calculating isotopic ratios. Reported results are the average of 3 replicate measurements of each sample, and errors are one standard deviation of the replicate analyses. Only data with relative standard deviations of less than 5% were reported; however, the average calculated percent relative standard deviations of the 101Ru∶104Ru and 102Ru∶104Ru ratios for all of the plotted samples were 1.60% and 1.48%, respectively. Finally, the triangular area in Fig. 3 indicates the zone of mixing between the respective ruthenium ratios for uranium-fission, plutonium-fission, and natural ruthenium. | ||
| Fig. 3 Ruthenium isotopic ratios of groundwater samples and vadose zone sediment extracts. | ||
| Sample identification | Depth/ft bgsa | Depth/m bgs | 101Ru∶104Ru (ratio) | Error (1 − σ) | 102Ru∶104Ru (ratio) | Error (1 − σ) |
|---|---|---|---|---|---|---|
| a Bgs indicates below ground surface. | ||||||
| 299-W11-25B | ||||||
| B1BWD1 | 260 | 79.2 | 1.934 | 0.0187 | 1.691 | 0.0210 |
| B1BWD4 | 279 | 85.0 | 1.722 | 0.0165 | 1.525 | 0.0240 |
| B1BWD7 | 301 | 91.7 | 1.743 | 0.0610 | 1.547 | 0.0371 |
| B1BWF0 | 321 | 97.8 | 1.720 | 0.0628 | 1.488 | 0.0198 |
| B1BWB7 | 340 | 103.6 | 1.686 | 0.0152 | 1.515 | 0.0440 |
| B1BWC2 | 360 | 109.7 | 1.743 | 0.0186 | 1.526 | 0.0157 |
| B1BWB8 | 380 | 115.8 | 1.691 | 0.0360 | 1.461 | 0.0554 |
| B1BWX7 | 409 | 124.7 | 1.562 | 0.0333 | 1.431 | 0.0169 |
| Shallow groundwater | ||||||
| 299-W11-39 | 243 | 74.1 | 1.959 | 0.0764 | 1.703 | 0.0903 |
| 299-W11-40 | 242 | 73.8 | 1.879 | 0.0618 | 1.661 | 0.0548 |
| 299-W11-41 | 244 | 74.4 | 1.913 | 0.0603 | 1.718 | 0.0514 |
| 299-W11-42 | 243 | 74.1 | 1.851 | 0.0711 | 1.644 | 0.0317 |
| Vadose zone sediment | ||||||
| C4104 | 47 | 14.3 | 1.992 | 0.0356 | 1.771 | 0.0337 |
| C4104-UFA | 64 | 19.5 | 1.930 | 0.0234 | 1.633 | 0.0222 |
| C4104 | 64 | 19.5 | 1.714 | 0.0291 | 1.525 | 0.0290 |
| C4104 | 76 | 23.2 | 1.753 | 0.0403 | 1.583 | 0.0253 |
| C4104-UFA | 94 | 28.7 | 1.984 | 0.0266 | 1.772 | 0.0113 |
| C4104 | 100 | 30.5 | 1.934 | 0.0077 | 1.710 | 0.0257 |
| C4104 | 106 | 32.3 | 1.982 | 0.0109 | 1.763 | 0.0069 |
| C4104-UFA | 116 | 35.4 | 1.967 | 0.0112 | 1.760 | 0.0071 |
| C4104 | 116 | 35.4 | 1.974 | 0.0089 | 1.771 | 0.0107 |
| C4104-UFA | 121 | 36.9 | 1.914 | 0.0228 | 1.730 | 0.0116 |
| C4104 | 127 | 38.7 | 1.957 | 0.0357 | 1.780 | 0.0265 |
| C4105-UFA | 70 | 21.3 | 1.966 | 0.0441 | 1.762 | 0.0429 |
| C4105 | 87 | 26.5 | 1.959 | 0.0159 | 1.768 | 0.0050 |
| C4105-UFA | 88 | 26.8 | 1.989 | 0.0157 | 1.778 | 0.0115 |
| C4105 | 88 | 26.8 | 1.967 | 0.0125 | 1.762 | 0.0066 |
| C4105-UFA | 93 | 28.3 | 1.984 | 0.0531 | 1.765 | 0.0312 |
| C4105 | 100 | 30.5 | 1.967 | 0.0204 | 1.760 | 0.0095 |
| C4105 | 103 | 31.4 | 1.974 | 0.0136 | 1.764 | 0.0074 |
| C4105-UFA | 107 | 32.6 | 1.972 | 0.0048 | 1.788 | 0.0076 |
| C4105 | 110 | 33.5 | 1.954 | 0.0131 | 1.758 | 0.0140 |
| C4105-UFA | 121 | 36.9 | 1.966 | 0.0257 | 1.764 | 0.0199 |
| C4105-UFA | 124 | 37.8 | 1.963 | 0.0189 | 1.771 | 0.0265 |
| C4105 | 130 | 39.6 | 1.959 | 0.0241 | 1.751 | 0.0041 |
As seen in Fig. 3, the 101Ru∶104Ru versus102Ru∶104Ru ratios for all of the samples fell along the mixing line between uranium and plutonium fission, indicating a negligible contribution from natural ruthenium. The standard deviations for the groundwater samples were generally larger than those for the vadose zone samples due to the lower concentrations of ruthenium present in the groundwater matrix. Ruthenium isotope ratios for most of the vadose zone samples collected near tank T-106, as well as the groundwater monitoring well network samples (collected at the top of the aquifer and specified as “shallow groundwater” in Fig. 3), clustered together. Additionally, these samples plotted closer to the uranium-fission end member for the 101Ru:104Ru versus102Ru:104Ru isotopic ratios, and the ratios were distinctly different from those of the deeper depth-discrete samples from well 299-W11-25B. Only the shallowest sample from well 299-W11-25B, which was collected near the water table, fell within the cluster of vadose zone and various shallow groundwater monitoring well (near water table) samples. The remaining samples (those collected below the top of the aquifer) from well 299-W11-25B had lower ratios and plotted closer towards the plutonium-fission end member.
The ruthenium ratios from extracts of two sediment samples from borehole C4104 plotted close to the deeper 299-W11-25B well water samples on the mixed fission line. These two lower ratio samples were collected in close proximity (depth-wise) to one another, at 64 and 76 ft (19.5–23.2 m) below ground surface (bgs), respectively. The sample collected from 64 ft (19.5 m) bgs was extracted using the 1∶1 sediment∶de-ionized water method and a separate aliquot was extracted using the UFA apparatus. Interestingly, the ruthenium isotopic ratios for these two extracts varied significantly. The water extract sample had a 101Ru∶104Ru ratio of 1.714 versus 1.930 for the UFA extracted sample. Likewise, the water extract sample had a 102Ru∶104Ru ratio of 1.525 versus 1.633 for the UFA extracted sample. Of all the concurrently water and UFA extracted samples, this one sample from 64 ft (19.5 m) bgs in borehole C4104 was the only sample to generate extracts with statistically significant differences in the isotopic ratios of 101Ru, 102Ru, and 104Ru. Based on these preliminary results, it appears that the statistically significant differences in ruthenium isotopic ratios from the sample collected from 64 ft (19.5 m) bgs in borehole C4104 are real. However, the focus of this paper was to document a quadrupole-based ICP-MS method capable of measuring ruthenium isotopic ratios; therefore, significantly more samples from all available discharge points (i.e., cribs and trenches) and known leak events (i.e., leaks from single-shell tanks and waste transfer pipelines) need to be evaluated prior to quantitative identification of contaminant source terms or leak events.
DRC method development
Although none of the samples analyzed as part of this study contained measurable amounts of natural ruthenium, an effort was made to determine the applicability of using the DRC to quantitate total ruthenium and determine natural versus fission-produced ruthenium. For this to occur, the concentration of stable ruthenium at a mass blocked by another stable isotope (i.e., 96Ru blocked by 96Mo) must be determined. Further, any other atomic interferences that could add to the signal at atomic mass unit 96, namely 96Mo and 96Zr, must be either removed or accounted for. Finally, the amount of natural ruthenium at isotopes 101, 102, and 104 can then be calculated based on each isotope’s natural relative abundance corrected by the concentration of 96Ru present in the sample.Method development was performed by analyzing a solution containing 1 ng ml−1 ruthenium, molybdenum, and zirconium in the DRC mode using oxygen as the reaction gas (Fig. 4). The data contained in Fig. 4 are the intensities (in counts per second) of the three elements, plotted on log scale, as a function of reaction gas flow-rate (varied from 0.1 to 1.2 ml min−1). A dramatic linear decrease in signal intensity, when the data are plotted on log scale, indicates that the element is highly reactive with the chosen reaction gas. Ideally, the analyte of interest will not be reactive while the unwanted elements with mass interferences will react quickly, preventing their injection into the mass analyzer quadrupole. As seen in Fig. 4, both molybdenum and zirconium were highly reactive with oxygen in the reaction cell. Conversely, although ruthenium experienced a drop in sensitivity with increasing reaction gas flow-rates, the phenomenon was related to dilution (fewer ions were injected into the mass analyzer quadrupole) and was not a result of the reaction of ruthenium with oxygen.
 | ||
| Fig. 4 Analysis of a solution containing 1 ng ml−1 ruthenium, molybdenum, and zirconium in the DRC mode using O2 as the reaction gas. Gas flow-rates were varied from 0.1 to 1.2 ml min−1. | ||
To demonstrate the application of this technique, a series of solutions containing 1 ng ml−1 total ruthenium with molybdenum and zirconium concentrations ranging from 0.01 to 100 ng ml−1 were analyzed in the DRC mode with an oxygen flow-rate of 0.8 ml min−1 (Table 4). Both zirconium and molybdenum were removed from solution in significant percentages for all of the samples run in DRC mode. In the sample containing 1 ng ml−1 total ruthenium with 100 ng ml−1 total zirconium and molybdenum, 99.9% of the zirconium and 99.7% of the molybdenum were removed at masses 90 and 95, respectively. There was a significant increase in the count per second intensity for all of the analytes as a function of increasing molybdenum and zirconium solution concentration. This result was an artifact of reduced reaction efficiency in the DRC due to the substantial increase in the number of ions in the cell. However, the effect of this increase in signal for non-reactive species, such as ruthenium, is negligible.
| Sample identification | 90Zr/cps | 95Mo/cps | 96Ru/cps | 101Ru/cps | 96Ru∶101Ru (ratio) | 96Ru∶101Ru∶correcteda (ratio) |
|---|---|---|---|---|---|---|
| a Indicates data was corrected for contribution from molybdenum and zirconium at atomic mass unit 96. | ||||||
| Blank solution | 1 | 2 | 1 | 1 | Not applicable | Not applicable |
| 1 ng ml−1 total Ru | 1 | 3 | 37 | 100 | 0.316 | 0.289 |
| 1 ng ml−1 total Ru + 0.01 ng ml−1 Mo and Zr | 1 | 3 | 44 | 118 | 0.317 | 0.294 |
| 1 ng ml−1 total Ru + 0.1 ng ml−1 Mo and Zr | 1 | 4 | 52 | 131 | 0.335 | 0.308 |
| 1 ng ml−1 total Ru + 0.5 ng ml−1 Mo and Zr | 1 | 10 | 64 | 151 | 0.368 | 0.308 |
| 1 ng ml−1 total Ru + 1 ng ml−1 Mo and Zr | 2 | 17 | 81 | 171 | 0.413 | 0.322 |
| 1 ng ml−1 total Ru + 5 ng ml−1 Mo and Zr | 4 | 74 | 191 | 298 | 0.575 | 0.342 |
| 1 ng ml−1 total Ru + 10 ng ml−1 Mo and Zr | 8 | 150 | 384 | 628 | 0.561 | 0.332 |
| 1 ng ml−1 total Ru + 25 ng ml−1 Mo and Zr | 16 | 365 | 757 | 1076 | 0.655 | 0.324 |
| 1 ng ml−1 total Ru + 50 ng ml−1 Mo and Zr | 27 | 721 | 1222 | 1316 | 0.864 | 0.330 |
| 1 ng ml−1 total Ru + 100 ng ml−1 Mo and Zr | 53 | 1460 | 2126 | 1541 | 1.30 | 0.365 |
| 100 ng ml−1 total Mo and Zr (standard mode) | 340000 | 583000 | ||||
| Ratio average | 0.630 | 0.321 | ||||
| Ratio standard deviation | 0.318 | 0.023 | ||||
| Percent relative standard deviation | 50.5 | 7.07 | ||||
Although the DRC was effective at removing nearly all of the contribution from zirconium and molybdenum at atomic mass unit 96, the accompanying reduction of signal for 96Ru did present quantitative issues. The total count rate associated with 96Ru for a solution containing 1 ng ml−1 total ruthenium was approximately 33 counts per second in the DRC mode; therefore, minor residual contributions from atomic interferences were important. Both 96Mo, with a natural abundance of 16.7%, and 96Zr, with a natural abundance of 2.8%, did impact the isotopic ratio of 96Ru:101Ru when the solution concentrations were increased above 0.5 ng ml−1 and 5 ng ml−1, respectively. To correct for this, the total counts at atomic mass unit 96 were corrected for the calculated contribution from 96Zr and 96Mo based on the 90Zr and 95Mo count rates. When this correction was performed, the average 96Ru:101Ru ratio for all 10 of the samples containing 1 ng ml−1 total ruthenium was 0.321, with a relative standard deviation of 7.07%. The calculated percent difference between the averaged 96Ru:101Ru ratio and the actual ratio based on natural abundance of ruthenium (0.325) was quite good (1.01%). Therefore, the DRC can be used to eliminate more than 99% of the contribution from 96Zr and 96Mo atomic interferences in combination with mass correction to quantitatively analyze samples containing up to 2 orders of magnitude more zirconium and molybdenum than ruthenium.
Conclusions
Precise ruthenium isotopic ratio measurements were achieved on a quadrupole-based ICP-MS using a low-flow Meinhard-type nebulizer and long sample acquisition times (150000 ms). Relative standard deviations were maintained at less than 0.5% when the total ruthenium solution concentration was 0.1 ng ml−1 or higher. Application of this method using groundwater samples and vadose zone sediment extracts from the Hanford Site indicated preliminarily that multiple sources (at least two) of contamination were present in the vadose zone and groundwater system near the T tank farm. Although this technique has yet to be widely applied to groundwater and sediment samples collected from the Hanford Site, it has the potential to be a powerful diagnostic tool to investigate contaminant sources. Environmental isotopic fractionation of ruthenium is assumed to be negligible so the isotopic ratios directly reflect source contributions. Source fission isotope variability is the result of variable contributions from plutonium versus uranium fission caused by a range in reactor power and exposure time.29Quantitative analysis of total ruthenium was made possible by analyzing the samples in the DRC mode using oxygen as the reaction gas. Nearly complete removal of molybdenum and zirconium atomic interferences was successful when these two elements were present at two orders of magnitude higher concentration than the ruthenium analyte concentration. These results were promising in that they could be used with stable-mass correction to quantitatively analyze samples containing mixed ruthenium (natural and fission-derived) and elements with overlapping atomic weights, and could eliminate the need to use conventional isotope-dilution techniques to quantify total ruthenium in aqueous samples.
Acknowledgements
The authors acknowledge F. J. Anderson and F. M. Mann at CH2M HILL Hanford Group, Inc. (Richland, WA) for providing project funding and technical guidance. The quality of the manuscript was greatly improved from review comments received from R. J. Serne (PNNL) and T. E. Jones (CH2M Hill Hanford Group, Inc.). Pacific Northwest National Laboratory is operated for the DOE by Battelle Memorial Institute under Contract DE AC05 76RL01830.References
- L. Y. Liang, B. H. Gu and X. P. Yin, Sep. Technol., 1996, 6, 111–122 CrossRef CAS.
- P. J. Kershaw, D. McCubbin and K. S. Leonard, Sci. Total Environ., 1999, 238, 119–132 CrossRef.
- M. McCartney, K. Rajendran, V. Olive, R. G. Busby and P. McDonald, J. Anal. At. Spectrom., 1999, 14, 1849–1852 RSC.
- S. Uchida, K. Tagami, W. Ruhm, M. Steiner and E. Wirth, Appl. Radiat. Isot., 2000, 53, 69–73 CrossRef CAS.
- J. K. Fredrickson, J. M. Zachara, D. W. Kennedy, R. K. Kukkadapu, J. P. McKinley, S. M. Heald, C. X. Liu and A. E. Plymale, Geochim. Cosmochim. Acta, 2004, 68, 3171–3187 CrossRef CAS.
- M. Dowdall, S. Gerland, M. Karcher, J. P. Gwynn, A. L. Rudjord and A. K. Kolstad, J. Environ. Radioact., 2005, 84, 111–130 CrossRef CAS.
- D. I. Kaplan and R. J. Serne, Radiochim. Acta, 1998, 81, 117–124 CAS.
- C. L. Liu, S. S. Li, X. Y. Wang, Z. M. Wang, C. T. Xin, H. F. Wang, R. J. Li, B. Li, L. T. Tang and L. Jiang, Radiochim. Acta, 2001, 89, 643–645 CrossRef CAS.
- EPA, Understanding Variation in Partition Coefficient, Kd, Values: Volume III. Review of Geochemistry and Available Kd Values for Americium, Arsenic, Curium, Iodine, Neptunium, Radium, and Technetium, EPA 402-R-04-002C, U.S. Environmental Protection Agency, Washington, D.C., 2004 Search PubMed.
- A. Mukherjee, A. E. Fryar and D. M. Lasage, Environ. Eng. Geosci., 2005, 11, 371–382 Search PubMed.
- W. W. Lukens, J. J. Bucher, D. K. Shuh and N. M. Edelstein, Environ. Sci. Technol., 2005, 39, 8064–8070 CrossRef CAS.
- W. Um and R. J. Serne, Radiochim. Acta, 2005, 93, 57–63 CrossRef CAS.
- M. J. Hartman, L. F. Morasch and W. D. Webber, Hanford Site Groundwater Monitoring for Fiscal Year 2004 PNNL-15070, Pacific Northwest National Laboratory, Richland, WA, 2005 Search PubMed.
- F. R. Chen, P. C. Burns and R. C. Ewing, J. Nucl. Mater., 2000, 278, 225–232 CrossRef CAS.
- K. Ollila, J. Nucl. Mater., 1992, 190, 70–77 CrossRef CAS.
- P. E. Dresel, J. C. Evans and O. T. Farmer, Investigation of Isotopic Signatures for Sources of Groundwater Contamination at the Hanford Site PNNL-13763, Pacific Northwest National Laboratory, Richland, WA, 2002 Search PubMed.
- H. Hidaka, P. Holliger and F. Gauthier-Lafaye, Chem. Geol., 1999, 155, 323–333 CrossRef CAS.
- R. J. Serne, B. N. Bjornstad, D. G. Horton, D. C. Lanigan, C. W. Lindenmeier, M. J. Lindberg, R. E. Clayton, V. L. LeGore, K. N. Geiszler, S. R. Baum, M. M. Valenta, I. V. Kutnyakov, T. S. Vickerman, R. D. Orr and C. F. Brown, Characterization of Vadose Zone Sediments Below the T Tank Farm: Boreholes C4104, C4105, 299-W10-196 and RCRA Borehole 299-W11-39 PNNL-14849, Pacific Northwest National Laboratory, Richland, WA, 2004 Search PubMed.
- M. Huang and A. Masuda, Anal. Chem., 1997, 69, 1135–1139 CrossRef CAS.
- H. Becker, C. Dalpe and R. J. Walker, Analyst, 2002, 127, 775–780 RSC.
- Y. V. Yi and A. Masuda, Anal. Chem., 1996, 68, 1444–1450 CrossRef CAS.
- X. X. Dai, C. Koeberl and H. Froschl, Anal. Chim. Acta, 2001, 436, 79–85 CrossRef CAS.
- I. Jarvis, M. M. Totland and K. E. Jarvis, Chem. Geol., 1997, 143, 27–42 CrossRef CAS.
- V. I. Baranov and S. D. Tanner, J. Anal. At. Spectrom., 1999, 14, 1133–1142 RSC.
- S. D. Tanner and V. I. Baranov, J. Am. Soc. Mass Spectrom., 1999, 10, 1083–1094 CrossRef.
- D. R. Bandura, V. I. Baranov and S. D. Tanner, J. Am. Soc. Mass Spectrom., 2002, 13, 1176–1185 CrossRef CAS.
- J. T. Rieger and M. J. Hartman, Fiscal Year 2005 Integrated Monitoring Plan for the Hanford Groundwater Performance Assessment Project PNNL-15176, Pacific Northwest National Laboratory, Richland, WA, 2005 Search PubMed.
- G. W. Thomas, in Methods of Soil Analysis: Part 3-Chemical Methods, ed. D. L. Sparks, Soil Science Society of America and the American Society of Agronomy, Madison, WI, 1996, vol. III, pp. 475–490 Search PubMed.
- R. A. Watrous and D. W. Wootan, Activity of Fuel Batches Processed through Hanford Separations Plants, 1944 through 1989 HNF-SD-WM-TI-794 Rev. 0, Lockheed Martin Hanford Corporation, Richland, WA, 1997 Search PubMed.
Footnotes |
| † Presented at 2006 Winter Conference on Plasma Spectrochemistry, Tucson, AZ, USA, January 8–14, 2006. |
| ‡ The T single-shell tank farm consists of twelve 500000 gallon and four 55000 gallon underground storage tanks built in 1943/44 and used for active storage of high-level nuclear waste until 1980. A number of these tanks are known to have leaked liquid wastes into the soil. |
| This journal is © The Royal Society of Chemistry 2006 |