Selenopeptide mapping in a selenium–yeast protein digest by parallel nanoHPLC-ICP-MS and nanoHPLC-electrospray-MS/MS after on-line preconcentration
Pierre Giustia, Dirk Schaumlöffela, Hugues Preud’hommea, Joanna Szpunara and Ryszard Lobinski*ab
aEquipe de Chimie Analytique Bio-inorganique, CNRS UMR 5034, Hélioparc, 2, av. Pr. Angot, F-64053 Pau, France
bDepartment of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664-Warsaw, Poland
First published on 29th November 2005
Abstract
ICP collision cell MS was optimized for the detection and retention-time marking of selenium-containing peptides in nanoHPLC (75 μm column) after on-line 100-fold preconcentration on a capillary (300 μm id) precolumn. The mobile phase composition, gradient and flow rate were chosen to allow electrospray-MS/MS to be successfully run in parallel in identical separation and preconcentration conditions in order to produce two matching sets of chromatograms: an element-specific one and a molecule-specific one. Knowledge of the retention time of a Se-containing peptide of interest allowed efficient data mining in the corresponding ES-MS chromatogram and the identification of minor Se-species. A third chromatogram was run to obtain collision-induced dissociation data for the target peptides. The performance of the method was demonstrated for a comprehensive on-line characterization of a mixture of peptides in a tryptic digest of a Se-containing protein fraction isolated by size-exclusion chromatography from a selenium yeast extract. The method allowed the identification of whole series of Se/S substitutions in individual peptides and, in some cases, sequencing of isomers differing in the position of selenomethionine residues in the amino acid sequence.
Introduction
The use of ICP-MS for element-specific detection in chromatography offers a number of incontestable advantages such as a femtogram level sensitivity, virtual independence of the signal intensity of the analyte’s molecular structure, absence of sample matrix suppression effects and a large linearity range.1–3 A major drawback of HPLC-ICP-MS is the impossibility of identification of the detected species without standards. Because the latter are rarely available in biochemistry, there is a growing interest in electrospray MS,4,5 and, recently, in MALDI MS6,7 for the identification of peaks in HPLC-ICP-MS chromatograms.In contrast to ICP-MS, the sensitivity of electrospray MS does critically depend on the molecular structure of the analyte and is suppressed by the sample matrix. It may also be considerably affected by the presence of an easily ionized compound in the analysed fraction, of which the ions can saturate the detector, especially when a TOF mass analyzer is used. Therefore the success of the ES-MS identification in the biochemical speciation analysis is critically dependent on the purity of an analyte arriving at a given moment at the ionization source.
The purification of element species prior to ES-MS has usually been carried out by multidimensional chromatography with the selection of fractions for analysis or a further purification on the basis of the total element concentration determined by ICP-MS.8 Analyses have been carried out off-line, which results in tedious procedures requiring a fairly large amount of sample. Large-bore columns have been used with their inherent drawbacks, such as low separation efficiency, large peaks and related sample dilution effects. The latter needed to be compensated by intermediate lyophilization steps which increased the complexity of procedures. The resulting relatively low purity of fractions has been responsible for the suppression of ionization and, consequently, for a 100–1000 discrepancy between the detection limits reported for ICP-MS and ES-MS/MS in speciation analysis.
The purity of an analyte arriving at a given moment at the detector can be improved by high resolution HPLC. NanoHPLC-electrospray Q-TOFMS, which allows the acquisition of full MS spectra with a 100 ms retention time resolution, is one of the basic techniques for peptide mapping in proteomics.9,10 For minor compounds the data interpretation remains, however, difficult. Recently McSheehy et al. addressed the challenge of identifying low abundance Se-peptides in protein digest of selenized yeast.11 Typical chromatograms were complex and difficult to interpret unless a database search was carried out for the, apparently more abundant, S-containing analogues of Se-peptides of interest. The fraction in which a S-containing peptide was identified was re-analysed, this time with forced fragmentation at the m + 48/z (48 is the mass difference between the most abundant 32S and 80Se isotopes). This approach required the prior knowledge of potentially present S-containing proteins and was limited to Se-peptides containing selenomethionine only, and that as a single residue.
Because a manual searching of the whole HPLC-ESMS chromatogram for minor compounds is difficult, the definition of areas of interest in a chromatogram, with a precision of few seconds, is becoming of paramount importance. It can be achieved by running HPLC-ICP-MS in exactly the same conditions as HPLC-ES-MS, either by splitting the effluent or by ensuring retention time reproducibility.12 The complementarity and synergy of ICP-MS and ES-MS/MS in speciation analysis have frequently been evoked but demonstrations of a combined on-line approach for biological samples have been rare. Wind et al.13,14 and, later, Pröfrock et al.15 reported an approach in which capillary (300 μm id) LC-ICP-MS, optimized for 31P detection, allowed the identification of phosphorylated peptides in a tryptic digest of model proteins prior to their structural analysis by the same chromatography technique with ES-QTOFMS detection. A similar approach was developed for S-containing protein standards.16 Our recent study demonstrated the parallel use of nanoHPLC-ICP-MS and ES-TOFMS for the determination of the tryptic digestion efficiency of a selenomethionyl calmodulin standard.17 The use of standard proteins in the above applications offered the advantage of injecting highly concentrated samples and ease of matching ICP-MS and ESMS peaks because of the simplicity of chromatograms.
The objective of this study was to investigate the potential of the parallel nanoHPLC-ICP-MS and QTOFMS detection for identifying heteroatom-containing peptides in a biological matrix. An on-line separation step was integrated into the protocol to allow the matrix removal, cleanup and de-salting of the sample and preconcentration of analytes. As a model system a selenized yeast protein digest studied earlier 6 by off-line fraction collection and MALDI MS and ES-MS was investigated.
Experimental
Apparatus
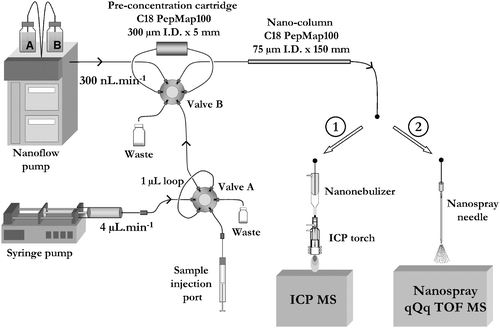 | ||
| Fig. 1 Instrumental setup of the two dimensional nanoHPLC with ICP MS (1) and ES-MS/MS detection. | ||
Reagents, solutions and materials
A selenized yeast sample, Sel-Plex (Alltech, Nicholasville, KY, USA), corresponding to a selenized yeast produced by a strain of Saccharomyces cerevisiae CNCM I-3060, batch ES-453, was used. Analytical reagent grade chemicals were purchased from Sigma–Aldrich (Saint-Quentin Fallavier, France) unless stated otherwise. Water (18.2 MΩ cm) was obtained from a Milli-Q system (Millipore, Bedford, MA). Trypsin solution (1.0 mg mL−1) was prepared by dissolving the enzyme (Trypsin Gold MS Grade, Promega, Madison, WI) in 50 mol L−1 acetic acid. A buffer solution (buffer A) containing 1 mmol L−1 CaCl2, 5 mmol L−1 2-mercaptoethanol and 50 mmol L−1 Tris was prepared and adjusted with HCl to pH 7.5.Microcentrifuge filters for ultrafiltration with a cut-off of 100 kDa were purchased from Millipore (Bedford, MA).
Procedures
| Valve position | Acetonitrile conc. (%) | ||||
|---|---|---|---|---|---|
| Time/min | A | B | Nanoflow pump (0.3 μl min−1) | Microflow pump (4 μl min−1) | |
| Preconcentration | 0 | Inject | Load | 2 | 1.6 |
| 4 | Inject | Inject | 2 | 1.6 | |
| NanoHPLC separation | 8 | Load | Inject | 2 | |
| 18 | Load | Inject | 50 | ||
| 20 | Load | Inject | 75 | ||
| 22.5 | Load | Inject | 95 | ||
| Cleaning | 25 | Load | Inject | 95 | |
| Re-equilibration | 26 | Load | Inject | 2 | |
Results and discussion
NanoLC-ICP-MS after on-line preconcentration step
A minimum concentration of acetonitrile of 1.6% was found to be necessary to wet the column stationary phase in order to enable interactions resulting in the sorption of hydrophobic peptides on a C18 reversed-phase cartridge. With these conditions the two first peptides in the digest, the highly hydrophilic XGHDQSGTK and XNAGR (where X denotes selenomethionine) (cf.ref. 6) were not retained. All the other peptides could be retained at a flow rate of 4 μL min−1 during a period of at least 4 min. This resulted in a sample solution load of 1 μl instead of 11 nl and hence in a 100-fold preconcentration factor. The recovery was verified by comparing a chromatogram obtained by nanoLC by direct 11 nL injection with one obtained for the same solution diluted 100 times and analysed by the setup shown in Fig. 1. The intensities of the peaks in the chromatograms were identical (within 10% uncertainty range) except for the above evoked first two peptides), which proves the quantitative recovery of the retained peptides.The nanoHPLC conditions were optimized in order to reduce the duration of the chromatogram and to improve the separation of the hydrophobic peptides. This is essential since the former work showed that many overlapping species could be present to a point to render a meaningful off-line ESMS/MS impossible.6 A nanoHPLC-ICP-MS chromatogram following on-line preconcentration, run under the optimum conditions, is shown in Fig. 2. It is very similar to that published elsewhere by large bore (4.6 mm) HPLC-ICP-MS.6 Except for the absence of the first two peaks evoked above, a major difference concerns the absence of a number of small peaks which could not be identified in the former study. This fact suggests that they are probably artifacts. Another difference is a better resolution of the most hydrophobic 1000–2000 Da peptides (peaks 7–18) in comparison with the former study.
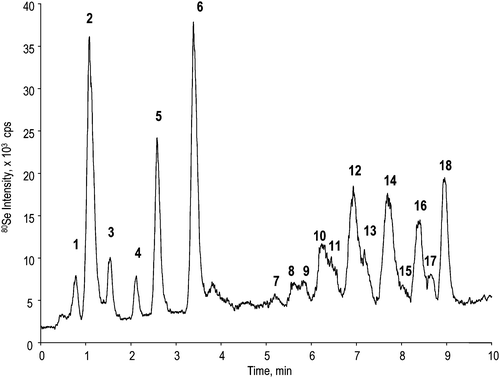 | ||
| Fig. 2 NanoHPLC-ICP-MS chromatogram. | ||
The 2-D (preconcentration–nanoHPLC) approach resulted in a considerable improvement in the reproducibility of the retention times in the chromatograms because of the elimination of a large part of the sample matrix during the preconcentration step. Indeed, whereas the average reproducibility of the peaks’ retention times using a direct 11 nL injection was ca. 8%, the use of the 2-D setup shown in Fig. 1 resulted in a 3% reproducibility.
NanoLC-ESMS after on-line preconcentration step
For the purpose of peak identification a total ion chromatogram (TIC) was acquired by nanoLC-ES-TOFMS in the identical preconcentration and chromatographic conditions as the HPLC-ICP-MS chromatogram shown in Fig. 2. A selenium isotopic pattern (corresponding to one or more selenium atoms, cf.ref. 19) was manually sought in the vicinity of the retention time of a Se-containing compound found in the nanoHPLC-ICP-MS chromatogram. A sulfur analogue was also sought at M − 48/z for the validation of the Se-isotopic pattern recognition. The extracted ion chromatograms (XICs), shown in Fig. 3 were subsequently plotted. The peak labels refer to the corresponding ICP-MS peaks. The mass spectra of a Se-containing peptide and of its sulfur analogue taken at the XIC apex are shown in the insets.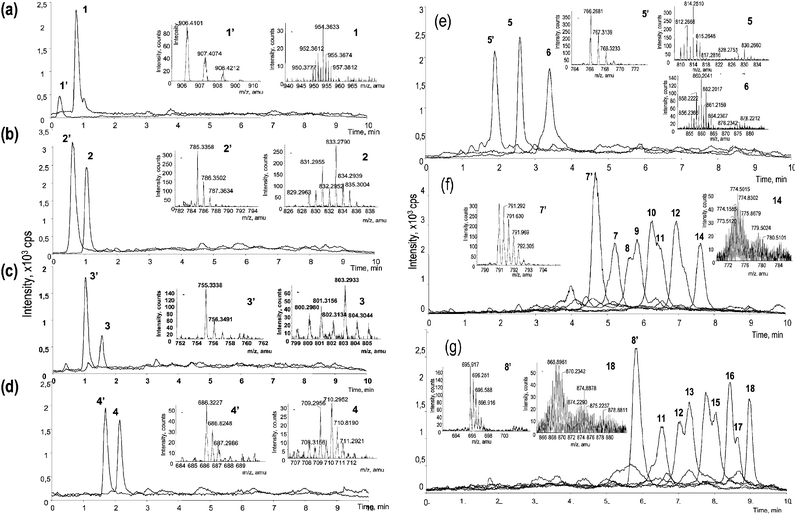 | ||
| Fig. 3 Extracted ion chromatograms (XIC) of selenium containing peptides and their sulfur-containing analogues in nanoHPLC-ESI-TOFMS. XIC mass window of 0.5 amu centered on the most intense peak of the isotopic pattern was used. The insets contain mass spectra taken at the peak apices for each Se-peptide and its sulfur analogue. The labels refer to peaks in the nanoHPLC-ICP-MS chromatogram (Fig. 2). The “prime” characters refer to the corresponding methionine-containing peptides (with an exception of peptide 5 containing both methionine and selenomethionine). | ||
The detection limits of electrospray MS were estimated by the signal-to-background ratio of the peaks in the mass spectrum corresponding to the 76Se and 77Se isotopes. Minor isotopes needed to be chosen because their detection in the mass spectrum was essential for the recognition of the Se isotopic pattern. These detection limits were compared with those obtained for the same peptides with ICP-MS calculated on the basis of the signal-to-background ratio in the chromatogram in Fig. 2 (ca. 100 fg as selenium). The ES-MS detection limits in the chromatographic mode (ca. 10 fmol per compound) are lower than in the off-line mode because at a given moment the Se-containing species is virtually the only one arriving at the detector. On the other hand, for ICP-MS the concentration detection limits are higher that in conventional 4.6 mm id HPLC because of the much lower injection volume (1 μl instead of 20–50 μl). When the facts that for the ICP-MS DL calculation the 80Se isotope with 49.6% abundance is used and for the ES-MS DL calculation the 76Se isotope with 7.6% abundance is used is taken into account, the sensitivities of nanoHPLC-ICP-MS and nanoES-TOFMS can be considered as similar. The optimized preconcentration/separation approach is therefore an ideal method to profit from the complementarity of ICP-MS and ES-MS detection in speciation analysis.
The ES-MS data for peaks 1–6 are straightforward to interpret. A selenium pattern can readily be recognized in the vicinity of the HPLC-ICP-MS apex. The selenium compound is preceded by a peak corresponding to the sulfur analogue which is more hydrophilic. Assuming a similar ionization, the concentrations of the corresponding Se- and S-peptides are fairly similar, taking into account that the most abundant S-isotope accounts for 95% of the element whereas that of Se only 49.6%. This finding contradicts a recent report11 which found distinctly higher concentrations of the S-analogues, to a point where they served to indicate the location of the possibly present minor Se-peptides.
Regarding peaks 5 and 6 in the nanoHPLC-ICP-MS chromatogram (Fig. 2), the ES-TOFMS data (Fig. 3(e)) demonstrate that the peaks correspond to the same peptide in which either one (peak 5) or both (peak 6) methionine residues were replaced by selenomethionine. The chromatogram shows that the SeMet/Met substitution increases the hydrophobicity of the peptide.
Electrospray TOFMS allows an elegant interpretation of the identity of peaks forming a late eluting group of peaks in the HPLC-ICP-MS chromatogram (peaks 7–18). The peptides expected in this range correspond to the largest digestion fragment of SIP18 protein—DDMNMDMGMGHDQSEGFGMK—in which one or more of the five methionine residues present were replaced by selenomethionine (Fig. 3(f)). Again, the replacement has a clear and regular effect on the hydrophobicity of the eluted peptide: the replacement of each sulfur atom by selenium increases the retention time of the peptide by ca. 45 s.
A comparison of the HPLC-ES-MS (XIC) chromatogram for these peptides with that obtained by HPLC-ICP-MS indicates the presence of compounds more hydrophilic than the Se-free DDMNMDMGMGHDQSEGFGMK peptide and, hence, belonging to other peptides. A careful examination of the ESMS data indicates the presence (Fig. 3(g)) of a series of peptides elongated on the N-terminus by two amino acid residues E and R. Glutamic acid and arginine are highly polar, which is responsible for the more hydrophilic character of these peptides. They originate from incomplete tryptic digestion failing to break the R–D bond.
The peaks in the XICs shown in Fig. 3(f) and 3(g) exhibit shoulders which are likely to be due to isomers with a different selenomethionine position. Indeed, the substitution of methionine by one or four SeMet residues in a DDMNMDMGMGHDQSEGFGMK peptide would lead to five isomers and the substitution of two or three methionines by SeMet to 10 isomers. A peptide with 5 methionine residues can therefore produce 31 different selenopeptides. An insight into the identity of isomers can be obtained by collision induced dissociation and tandem MS.
NanoLC-ES-MS/MS after on-line preconcentration step
Although the accurate (±20 ppm on the average) m/z determination can enable, in some cases, the identification of peptides and proteins in databases, CID-MS offers a more elegant and complete approach for this purpose. In the chromatographic mode the acquisition of a tandem mass spectrum is usually triggered off for a peak exceeding a certain intensity threshold. In the analysis of a yeast extract for minor compounds it is therefore unlikely that the fragmentation of all the compounds of interest will be automatically triggered off. McSheehy et al. proposed to use for data dependent acquisition (DDA) the sulfur analogues because of their much higher intensity.11 However, as discussed above, the S-analogues were not necessarily more intense and a number of S-containing proteins cannot be selenized at all. Another disadvantage of the DDA is that during the time of the MS/MS acquisition no detection of eluting peptides is possible.Therefore it was decided to run an additional chromatogram, this time with forced fragmentation of Se-containing ions. The CID mass spectra allowed the attribution of a sequence to the Se-peptides found. These sequences, together with the identity of the protein found, are shown in Table 2. By this approach all the peaks (except peak 1) in the nanoHPLC-ICP-MS chromatogram could be identified.
| Peak | Peptide sequenceab | MW [M + H]+c theoretical/experimental | ΔM (ppm) | Protein |
|---|---|---|---|---|
| a Non-methionine peptides allowing the confirmation of the protein identity included: LQGNDDSHQK (SIP18), GIANDWK (SIP18), EYITDK (HSP12), ASEALKPDSQK (HSP12), GVFGVHDSAEK (HSP12), GFGEK (HSP12), VHGEEDPTKK (HSP12), DNAEGQGESLADQAR (HSP12), GKDNAEGQGESLADQAR (HSP13), SSIGSQDSSDVEDVK (YMZ 7).b Peptides indicating the presence of other proteins: SVVIHAGQDDLGKGDTEESLK (superhydroxide dismutase), TGNAGPRPACGVIGLTN (superhydroxide dismutase), NQILVSGEIPSTLNEESKDK (HSP26), VITLPDYPGVDADNIK (HSP26), FAEQYSDAAFYK (thioredoxin II), THQQENLQSMR (thioredoxin II).c Molecular mass of the most abundant ion. | ||||
| 2′ | TYENMK | 785.350/785.343 | −9 | SIP18 osmotic stress |
| 2 | TYEN![[X with combining low line]](https://www.rsc.org/images/entities/b_char_0058_0332.gif) K K | 833.295/833.293 | −2 | |
| 5′ | SNMMNK | 766.322/766.307 | −20 | |
| 5 | SNMNK | 814.267/814.267 | 0 | |
| 5 | SNMNK | 814.267/814.267 | 0 | |
| 6 | SNNK | 862.212/862.215 | 4 | |
| 7′ | ERDDMNMDMGMGHDQSEGGMK | 2371.890/2371.923 | 14 | |
| 7 | 1 (ERDDMNMDMGMGHDQSEGGMK) | 2418.834/ 2418.873 | 16 | |
| 8–9 | 2 (ERDDMNMDMGMGHDQSEGGMK) | 2466.780/2466.831 | 21 | |
| 10–11 | 3 (ERDDMNMDMGMGHDQSEGGMK) | 2512.725/2512.758 | 13 | |
| 12 | 4 (ERDDMNMDMGMGHDQSEGGMK) | 2560.671/2560.722 | 20 | |
| 14 | ERDDNDGGHDQSEGGK | 2606.616/2606.640 | 9 | |
| 8′ | DDMNMDMGMGHDQSEGGMK | 2086.747/ 2086.749 | 1 | |
| 11 | 1 (DDMNMDMGMGHDQSEGGMK) | 2133.690/2133.684 | −2 | |
| 12–13 | 2 (DDMNMDMGMGHDQSEGGMK) | 2181.635/2181.636 | 1 | |
| 15 | 3 (DDMNMDMGMGHDQSEGGMK) | 2227.581/2227.611 | 14 | |
| 16–17 | 4 (DDMNMDMGMGHDQSEGGMK) | 2275.526/2275.554 | 12 | |
| 17 | DDNDGGHDQSEGGK | 2321.472/2321.472 | 0 | |
| 3′ | DYMGAAK | 755.339/ 755.333 | −8 | HSP12 glucose and lipid |
| 3 | DYGAAK | 803.284/803.293 | 11 | Regulator |
| 4′ | T HQQENLQSMR | 1371.643/1371.640 | −2 | YMZ 7 |
| 4 | T HQQENLQSR | 1419.589/1419.600 | −8 | |
Because of the improved purity of the compounds arriving at the detector the on-line approach developed is clearly superior to the off-line approach reported elsewhere.6 In particular, it allowed a detailed characterization of the most hydrophobic peptides (peaks 7–18 in Fig. 2) for which the off-line ES-MS/MS failed. Also, Se-containing peaks from another protein (YMZ7) were identified.
CID-MS allows one to distinguish between compounds with the same mass but differing in the position of selenomethionine. Fig. 4 illustrates this for the peptide SNMMNK, which contains two selenomethionine residues. The zoom of the CID mass spectrum shows the y fragments characteristic of different positions of selenomethionine in the sequence.
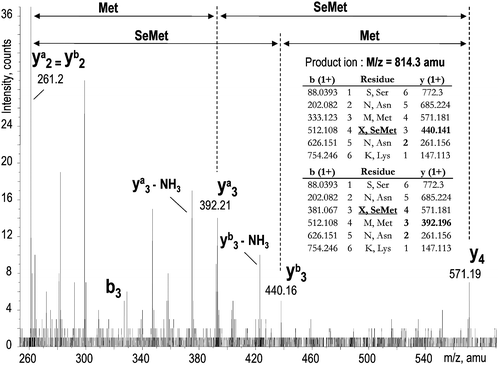 | ||
| Fig. 4 An MS/MS spectrum demonstrating the presence of two isomers with a different position of selenomethionine in the sequence (peak 5 in Fig. 2). | ||
A limitation to identifying the isomers is the default software setting which prevents the repeated fragmenting of the ion of the same mass during 45 s. Because the isomers have the same mass and elute closely to each other, only their first elution is fragmented to provide the sequence information.
Conclusion
On-line preconcentration on a C18 capillary precolumn allows salt removal and sample cleanup, which results in an improvement of retention time reproducibility and assures close to ideal detection conditions of selenium species by electrospray MS/MS. The parallel ICP-MS detection makes it possible to determine in a few-seconds large regions in which Se-containing peaks occur and facilitates finding them in the corresponding ESMS spectra. In these conditions the detection limits of ESMS and ICPMS are comparable within a factor of 5, which makes the combined approach valuable for speciation analysis. Another advantage is a 100-fold increase in the injection volume which alleviates the principal limitation of nanoHPLC in trace speciation analysis, i.e., the small sample volume allowed. The method is as efficient as the previously published off-line approach by large bore HPLC with fraction collection and MALDI and ESMS6 but is more sensitive, less time consuming and definitely more elegant. It opens the way to high throughput data acquisition on selenoproteomes of biological organisms.Acknowledgements
The study was carried out in the frame of the ACI CNRS (New Analytical Methods and Sensors) programme. The Aquitaine Region is acknowledged for the support of instrumentation via CPER 20.6 and 21.6. The authors thank Dr. Mihaly Dernovics for his help in sample preparation.References
- A. Sanz-Medel, M. Montes-Bayon and M. L. Fernandez Sanchez, Anal. Bioanal. Chem., 2003, 377, 236 CrossRef CAS.
- M. Montes-Bayon, K. DeNicola and J. A. Caruso, J. Chromatogr. A, 2003, 1000, 457 Search PubMed.
- J. Szpunar, R. Lobinski and A. Prange, Appl. Spectrosc., 2003, 57, 102A CrossRef CAS.
- H. Chassaigne, V. Vacchina and R. Lobinski, Trends Anal. Chem., 2000, 19, 300 CrossRef CAS.
- E. Rosenberg, J. Chromatogr. A, 2003, 1000, 841 Search PubMed.
- J. R. Encinar, L. Ouerdane, W. Buchmann, J. Tortajada, R. Lobinski and J. Szpunar, Anal. Chem., 2003, 75, 3765 CrossRef.
- J. Ruiz Encinar, A. Polatajko, R. Lobinski and J. Szpunar, Analyst, 2004, 129, 846 RSC.
- J. Szpunar and R. Lobinski, Anal. Bioanal. Chem., 2002, 373, 404 CrossRef CAS.
- Y. Shen and R. D. Smith, Electrophoresis, 2002, 23, 3106 CrossRef CAS.
- H. Wang and S. Hanash, J. Chromatogr., B, 2003, 787, 11 CrossRef CAS.
- S. McSheehy, J. Kelly, L. Tessier and Z. Mester, Analyst, 2005, 130, 35 RSC.
- M. Wind and W. D. Lehmann, J. Anal. At. Spectrom., 2004, 19, 20 RSC.
- M. Wind, M. Edler, N. Jakubowski, M. Linscheid, H. Wesch and W. D. Lehmann, Anal. Chem., 2001, 73, 29 CrossRef CAS.
- M. Wind, D. Gosenca, D. Kubler and W. D. Lehmann, Anal. Biochem., 2003, 317, 26 CrossRef CAS.
- D. Pröfrock, P. Leonhard, W. Ruck and A. Prange, Anal. Bioanal. Chem., 2005, 381, 194 CrossRef CAS.
- M. Wind, A. Wegener, A. Eisenmenger, R. Kellner and W. D. Lehmann, Angew. Chem. Int. Ed. Engl., 2003, 42, 3425 CrossRef CAS.
- P. Giusti, D. Schaumlöffel, J. Ruiz Encinar and J. Szpunar, J. Anal. At. Spectrom., 2005, 20, 1101 RSC.
- D. Schaumlöffel, P. Giusti, J. Szpunar and R. Lobinski, French Patent Application FR 05 05884, June 2005.
- J. R. Encinar, M. Sliwka-Kaszynska, A. Polatajko, V. Vacchina and J. Szpunar, Anal. Chim. Acta, 2003, 500, 171 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |