Silicate digestion with fructose under mild conditions
Gang
Lu
a,
Jonathan E.
Grossman
a,
Joseph B.
Lambert
*a,
Zhijun
Xiao
b and
Dan
Fu
b
aDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA. E-mail: jlambert@northwestern.edu
bSchlumberger Corp., Oilfield Chemical Products, 110 Schlumberger Drive, MD-3, Sugar Land, TX 77478, USA
First published on 19th May 2006
Abstract
Treatment of silica gel or silicate mineral with an aqueous solution of the weak base triethylenetetraamine and the sugar fructose results in partial dispersion into particles smaller than 450 nm under conditions of ambient pressure and mild heating (60–100 °C) in times less than 1 h. Redeposition occurs at longer times. The final material has 13–31% higher surface area and 17–18% lower D50 (50% of the accumulated weight percentage is smaller than this particle size). Dissolution or decrystallization of some of the aluminosilicate phase is indicated by X-ray diffraction for the Berea sandstone. The proposed mechanism involves deprotonation at the anomeric hydroxyl, followed by attack on the solid silicate centers by fructose to form a pentacoordinate silicon complex. The stereochemistry of fructose then allows a planar diolato ring to form containing C2 and C3 of fructose and a second position on pentacoordinate silicon. The reaction results in displacement of a portion of the silicate matrix. Reformation of Si–O–Si bonds gives rise to redeposition. An equilibrium between dissolution and redeposition results in the final reworked material with reduced particle size.
Introduction
Silicates constitute the largest mineral class in the earth's lithosphere. Because petroleum often is found embedded in silicate formations, the process of petroleum extraction requires dissolution, digestion, pulverization, or otherwise loosening the solid matrix at the bottom of a well to permit flow of the desired material.1 Current technology involves pumping hydrogen fluoride (HF) to the silicate formations. Production and handling of HF occurs on site, with potential danger both to technicians and to the environment. The development of alternative strategies to dissolve silicate minerals therefore is important to the petroleum industry and for the preservation of the environment.Dissolution of silicates also is desirable or necessary in several other contexts. (1) The construction industry often is faced with demolition of concrete structures. Removal and transport of a dissolved or slurried silicate would be far easier than of solid concrete blocks.2 (2) Solubilized silicate minerals are an important starting material for the silicon and organosilicon industries when economically viable.3 (3) Rice hulls, containing ca. 20% silica, are a potential raw material for silicon industries but again must be solubilized.4 In addition, marine organisms such as diatoms, sponges, and radiolarians metabolize silica to construct much of their structure. Silicate minerals thus are an important biosource, although the biochemical mechanisms are not fully understood.
Several groups have reported the dissolution of silica in basic, aqueous media to produce anionic alkoxy and aryloxy silicates.5–12 Others have treated aqueous silicic acid with polyols to form penta- and hexacoordinated polyolate complexes.13–16 We recently reported that silicic acid forms stable, hypercoordinated silicates with sugars,17 and Klüfers et al. reported the X-ray structures of complexes between arylsiliconates and sugars.18
Laine and co-workers19–24 found that the reaction of silica gel, fused silica, or sand at high temperatures with ethylene glycol and group 1 metals or group 2 metal oxides produced penta- and hexacoordinated glycolato silicate complexes. In a patent, Bailey et al. described the reaction of silica in mixed solvents of simple alcohols (ethanol or propanol) and aromatics (benzene or xylene) with strong base to produce alkoxysilanes.23 Suzuki and co-workers demonstrated that gaseous dimethyl or diethyl carbonate reacts with alkali-treated silica at very high temperatures to form alkoxysilanes in high yield.24 These routes require strong bases and temperatures usually in excess of 200 °C for a prolonged period of time.
The use of HF or extremely strong bases is not optimal from many points of view. We describe herein a novel and simple procedure for digestion of silicate materials utilizing the weak base triethylenetetraamine (TETA) instead of strong bases. The procedure is carried out at lower temperatures than most previous dissolution procedures. This study was inspired in part by the work of Laine et al.20 and the observation of Frye et al. that spirosilicates react with methanol and amines to form pentacoordinated silicates.25 Our work, however, finds that TETA alone is relatively inactive and needs the addition of a chelating agent, which evolved from our previous work with sugar silicates.17
Results
Digestion experiments were carried out on two silicate materials: silica gel (pore size 60 Å) and Berea sandstone (ground to a fine powder). All experiments were carried out in aqueous solution containing one or more of the following components: TETA to serve as a weak base, EDTA to complex metal cations, and D-fructose to chelate with silicate oxygens. Both temperature and pressure were varied. The weight of the silicate material was measured initially and after treatment. In addition in one set of experiments as indicated, the dissolved silicate levels were measured as a function of time by inductively coupled plasma (ICP).Silica gel at ambient pressure
Treatment of silica gel in an open round-bottomed flask at 60 °C for 1 h in the presence of TETA, EDTA, and fructose led to a 36% loss of mass. Similar treatment at 80 °C for 48 min led to a 40% loss of mass. Conditions identical to the second experiment but without fructose led to only a 10% loss of mass.Berea sandstone at ambient pressure
Similar treatment of powdered Berea sandstone with all three components at 60 °C for 4 h led to a 33% loss of mass. An experiment at 80 °C for 48 min led to 32% loss of mass. Three further experiments under the latter conditions were performed without one of the components. When fructose and TETA were present but EDTA was not, loss of mass was 28% after 48 min. When EDTA and TETA were present but fructose was not, there was no loss of mass. When EDTA and fructose were present but TETA was not, there was no loss of mass. Mass loss was measured as a function of time in a series of discrete experiments at ambient pressure and 100 °C (Fig. 1). After 30 min a mass loss of only 5% was obtained. The loss increased to 38% at 48 min and 34% at 1 h. After an hour, secondary precipitation26 of silicate occurred, resulting in little or no net mass loss. At a slightly lower temperature, similar results were obtained (Fig. 2). | ||
| Fig. 1 Dissolution of Berea sandstone at ambient pressure (100 °C). | ||
 | ||
| Fig. 2 Dissolution of Berea sandstone at 80 °C and ambient pressure. | ||
Berea sandstone under pressure
Treatment of powdered Berea sandstone in an autoclave permitted use of higher temperature (150 °C) and pressure. With all three components present, loss of mass as a function of time was monitored in a series of discrete experiments, as illustrated in Fig. 3. After only 1 h, a mass loss of 39% was achieved. With prolonged heating, however, mass loss decreased drastically, leveling off at about 12% after 7 h. Secondary precipitation of dissolved silicates apparently was occurring. | ||
| Fig. 3 Dissolution of Berea sandstone in an autoclave at 150 °C. | ||
Effect of pore size on filtration
All the above experiments were carried out with filtration of the aqueous solution through a 0.45 µm (450 nm) filter. The weights reflect the amount of material that failed to penetrate the filter. When a 0.2 µm (200 nm) filter was used, little or no mass loss was observed (and only trace amounts of silicate were detected by ICP analysis of the filtrate).Powder X-ray diffraction
Untreated samples and samples treated with TETA and fructose for 60 min at 60 °C were examined for their diffraction properties. Silica gel samples showed no diffraction peaks other than those of the sample holder and consequently are amorphous. Untreated Berea sandstone showed strong quartz peaks and minor aluminosilicate phases (clays and feldspars). Sandstone recovered after treatment contained less aluminosilicates than untreated material.Particle size distribution
Untreated samples and samples treated as for the X-ray experiments were analyzed for particle size distribution. In a plot of volume% vs. particle size, untreated silica gel exhibited one major peak with a maximum at ca. 75 µm and a smaller peak with a maximum at ca. 5 µm. The overall specific surface area was 0.182 m2 g−1. The respective values of D10, D50, and D90 were 37.1, 67.9, and 107.4 µm (10, 50, and 90% of the accumulated weight percentage are smaller than these respective values). The volume weighted mean was 107.4 µm. Following treatment of the silica gel, the two original peaks, now with maxima at ca. 8 and ca. 63 µm, were joined by a small new peak with a maximum at ca. 450 µm. The overall specific surface area had increased to 0.239 m2 g−1, the respective values of D10, D50, and D90 had decreased to 14.6, 56.4, and 106.8 µm, and the volume weighted mean had decreased to 106.8 µm.The same plot for untreated Berea sandstone (Fig. 4) showed one major peak with a maximum at ca. 160 µm and two broad minor peaks with maxima at ca. 6 and ca. 20 µm with an overall specific surface area of 0.196 m2 g−1. The respective values of D10, D50, and D90 were 22.9, 144.9, and 271.0 µm, and the volume weighted mean was 271.0 µm. Following treatment of the Berea sandstone, the peak at lowest particle size had become a shoulder (ca. 5–10 µm), the second small peak now with a maximum at ca. 25 µm had increased slightly in size, and the large peak had moved only slightly to a maximum at ca. 145 µm. The specific surface area had become 0.221 m2 g−1, D10/D50/D90 were 18.0/119.0/222.0 µm, and the volume weighted mean 122.5 µm.
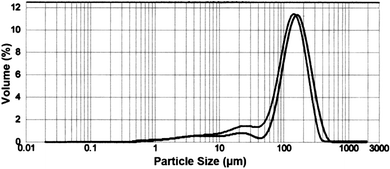 | ||
| Fig. 4 Particle size distribution as a function of percentage volume for (right curve) untreated Berea sandstone and (left curve) treated Berea sandstone. | ||
Discussion
In past work, silicate has been digested under highly basic or acidic conditions.22,26 With a weak base such as TETA, very little dissolution or digestion occurs. Silica gel treated at 80 °C and atmospheric pressure for 48 min showed a mass loss of only 10% in the presence of TETA alone. With both TETA and fructose present under the same conditions, the mass loss was 40%. Clearly, fructose plays a critical role in optimizing digestion. At only 60 °C for 60 min, with both TETA and fructose, the mass loss was 35%. EDTA was present in all these experiments, and mass loss was measured by weighing the solid material before and after treatment.Treatment of Berea sandstone with TETA, fructose, and EDTA at 60–80 °C for 48–240 min led to a mass loss of 32–33%. When EDTA was omitted from the experiment, there still was 28% mass loss after 48 min. When either TETA or fructose was omitted, however, the mass loss was zero after 48 min. Thus EDTA plays a minor role, but both the chelating agent fructose and the weak base TETA are necessary for any dissolution to occur. As with silica gel, the mass loss in these experiments with Berea sandstone was measured by measuring the weight of the solid before and after treatment.
At slightly higher temperatures (100 °C), mass loss was obtained as a function of time in a series of discrete experiments. Fig. 1 shows that the maximal mass loss occurred at 48–60 min under these conditions. This observation indicates secondary precipitation, which has been observed widely.26 Treatment with TETA and fructose gives optimal results after a relatively short amount of time. Thus apparently negative results (no mass loss) could occur if experiments were allowed to run for longer periods of time. Similar results were obtained at 80 °C (Fig. 2). Although ammonium ions can serve as templates for silicate construction, our basic conditions prohibit the formation of ammonium ions from TETA.
The maximal allowable temperature with ambient pressure is approximately 100 °C. Higher temperatures would require higher pressures, which we achieved by the use of an autoclave. With TETA, fructose, and EDTA present at 150 °C for 7 h in an autoclave, mass loss was monitored as a function of time (Fig. 3). After 1 h, mass loss was 39%, comparable to results at lower temperatures and pressures. After 5 h, however, mass loss dropped to about 12% and remained there.
All these experiments involved filtering the solution through a 0.45 µm filter and weighing the mass of the material that failed to pass through the filter. When a 0.2 µm filter was used, in the case of reaction at 80 °C for 4 h, little mass loss was observed. Thus the digestion involves reduction in particle size from macroscopic to nanometer (200–450 nm). A 200 nm sized particle corresponds to an –(O–Si)n– length with n equaling approximately 400 in one dimension (each O–Si–O unit requires about 0.25 nm). The dissolution conditions have reduced the silicate minerals from an essentially infinite solid polymer to a highly disperse, low molecular weight material that passes through a 0.45 µm filter but fails to pass through a 0.20 µm filter.
The X-ray experiments indicated that the silica gel samples were amorphous and the Berea sandstone samples were crystalline, even after treatment. No further information could be obtained from the silica gel samples. Diminution of the aluminosilicate peaks in relation to the quartz peaks for the treated Berea sandstone samples indicates preferential digestion of the aluminosilicate phases. Such phases have higher surface area and looser crystal structure than the quartz phase and hence are more susceptible to digestion.
Measurement of particle size distributions revealed that profound changes had occurred following treatment of both materials. The specific surface area of the silica gel had increased by 31%, indicating the more exposed nature of the treated material. Consequently, the volume weighted mean had decreased by 5%. This overall figure, however, is not descriptive of the changes that had occurred within the sample. Although a small third peak appeared at larger size, the maximum of the major peak moved to a smaller particle size. The net result is a 60% decrease in D10 and a 17% decrease in D50, but only a 0.5% decrease of D90. Treatment of silica gel by TETA and fructose has increased the surface area and decreased the particle size.
Treatment of Berea sandstone does not give rise to the third peak, but similar results are obtained for bulk properties. The plot in Fig. 4 shows that the major peak moves to lower particle size after treatment, and the minor peak at lower particle size increases in intensity. The overall specific surface area increased by 13%, and the volume weighted mean decreased by 20%. Without the new peak at larger size, D10, D50, and D90 all decreased, respectively by 22, 18, and 18%. Once again, treatment by TETA and fructose has increased the surface area and decreased the particle size.
Scheme 1, based on our earlier observations,17 provides a possible mechanism for the action of fructose on silicate in a weakly basic aqueous solution. In the first step, the weak base TETA removes the most acidic hydrogen, which is found at the anomeric position of fructose. In the second step, the basic, anomeric oxygen attacks a tetracoordinate silicate grouping (the wiggly lines in Scheme 1 indicate further connections to silicon within the solid silicate matrix). The result, following our experiments on the reaction between fructose and silicic acid under highly basic conditions,17 is formation of pentacoordinate silicon.
 | ||
| Scheme 1 | ||
It is likely that this charged material would tend to be more soluble in water than the uncharged tetracoordinate silicate. Such a step by itself, however, cannot result in dissolution, as no portion of the silicate matrix has split off. Otherwise, TETA alone should bring about dissolution, which it does not. There must be a special role for fructose, which we envisage to be analogous to its reaction with silicic acid [Si(OH)4].17 We observed that fructose, and only certain other furanose (five-membered-ring) sugars, react with silicic acid, because they have an anomeric HO group (at the 2 position in fructose) that is cis to an adjacent HO group (at the 3 position). Only when this structural grouping (cis HO–C–C–OH) is present can a nearly planar chelate five-membered diolato ring form. When the adjacent HO group is trans or when the sugar ring has six members (pyranose), formation of the planar diolato ring is stereochemically impossible.17
In the present context, the 3-OH in fructose displaces a silicate O–Si bond in the third step of Scheme 1 to form the illustrated, planar diolato chelate product and an extruded silicate fragment (“HO–”). This formal step can occur by either an associative or a dissociative mechanism, and at present we cannot choose between the two possibilities. In the associative mechanism, TETA initially removes the proton from the 3-OH group. The resulting alkoxide ion attacks silicon to form a doubly charged hexacoordinated chelate (the planar diolato ring). Our earlier experiments17 and those of Kinrade et al.13–16 have demonstrated that hexacoordinate silicon can occur under these conditions. Loss of siloxide then restores pentacoordination and produces (after reprotonation) the extruded silicate fragment “HO–” depicted as a final product in Scheme 1. In the dissociative mechanism, loss of the siloxide fragment occurs initially from the pentacoordinated species to form tetracoordinate silicon. Attack by the deprotonated 3-OH group of fructose then produces the pentacoordinate product in Scheme 1. By either variant, these final steps begin the process of dissolution. Repetition of the mechanism of Scheme 1 at multiple sites in the solid continues to degrade the silicate matrix. We have found that fructose succeeds in breaking the matrix down in this fashion to particles of the size 200–450 nm.
Redeposition has been discussed thoroughly in the literature.26 In the present context it represents the reversal of the process described in Scheme 1, probably without the involvement of fructose. Redeposition or polysilicate formation takes place by reformation of the stable Si–O–Si bonds. Thus dissolution and redeposition are two aspects of an equilibrium process. The redeposited material of course is not the same as the original. Treatment of silica gel or Berea sandstone with TETA and fructose results in some dissolution of the aluminosilicate phases, some dispersion to colloidal-like particles of size 200–450 nm, overall reduction in particle size, and some production of redeposited solid material. This result, achieved by environmentally benign conditions of weak basicity, would permit flow of petroleum from silicate formations or slurrying of concrete residues.
Experimental
Materials
Berea sandstone core (quartz content >85%) was supplied by Schlumberger Corp. and was ground to a fine powder of size <200 mesh ASTM (74 µm) with a mortar and pestle. Silica gel for column chromatography (pore size 60 Å) was purchased from Fisher and used as received.Procedures
Acknowledgements
This work was supported by the National Science Foundation (Grant no. CHE-0349412) and the Schlumberger Corporation.References
- C. Crowe, J. Masmonteil and R. Thomas, Oilfield Rev., 1992, 4, 24–40 Search PubMed.
- C. Müller, http://www.b-i-m.de/Public/IBAC/mueller.htm..
- R. M. Laine, K. Y. Blohowiak, T. R. Robinson, M. L. Hoppe, P. Nardi, J. Kampf and J. Uhm, Nature, 1991, 353, 642–644 CrossRef CAS.
- M. Z. Asunción, I. Hasegawa, J. W. Kampf and R. M. Laine, J. Mater. Chem., 2005, 15, 2114–2121 RSC.
- D. W. Barnum, Inorg. Chem., 1970, 9, 1942–1943 CrossRef CAS.
- D. W. Barnum, Inorg. Chem., 1972, 11, 1424–1429 CrossRef CAS.
- F. P. Boer, J. J. Flynn and J. W. Turley, J. Am. Chem. Soc., 1968, 90, 6973–6977 CrossRef CAS.
- A. Boudin, G. Cerveau, C. Chuit, R. J. P. Corriu and C. Reye, Angew. Chem., Int. Ed. Engl., 1986, 25, 473–474 CrossRef.
- A. Boudin, G. Cerveau, C. Chuit, R. J. P. Corriu and C. Reye, Organometallics, 1988, 7, 1165–1171 CrossRef CAS.
- J. J. Flynn and F. P. Boer, J. Am. Chem. Soc., 1969, 91, 5756–5761 CrossRef CAS.
- C. L. Frye, J. Am. Chem. Soc., 1964, 86, 3170–3171 CrossRef CAS.
- A. Weiss, G. Reiff and A. Weiss, Z. Anorg. Allg. Chem., 1961, 311, 151–179 CrossRef CAS.
- S. D. Kinrade, J. W. Del Nin, A. S. Schach, T. A. Sloan, K. L. Wilson and C. T. G. Knight, Science, 1999, 285, 1542–1545 CrossRef CAS.
- S. D. Kinrade, E. W. Deguns, A. M. E. Gillson and C. T. G. Knight, Dalton Trans., 2003, 3713–3716 RSC.
- S. D. Kinrade, K. J. Maa, A. S. Schach, T. A. Sloan and C. T. B. Knight, J. Chem. Soc., Dalton Trans., 1999, 3149–3150 RSC.
- S. D. Kinrade, A. S. Schach, R. J. Hamilton and C. T. G. Knight, Chem. Commun., 2001, 1564–1565 RSC.
- J. B. Lambert, G. Lu, S. R. Singer and V. M. Kolb, J. Am. Chem. Soc., 2004, 126, 9611–9625 CrossRef CAS.
- P. Klüfers, F. Kopp and M. Vogt, Chem.–Eur. J., 2004, 10, 4538–4545 CrossRef.
- K. Y. Blohowiak, D. R. Treadwell, B. L. Mueller, M. L. Hoppe, S. Jouppi, P. Pansal, K. W. Chew, C. L. S. Scotto, F. Babonneau, J. Kampf and R. M. Laine, Chem. Mater., 1994, 6, 2177–2192 CrossRef CAS.
- H. Q. Cheng, R. Tamaki, R. M. Laine, F. Babonneau, Y. Chujo and D. R. Treadwell, J. Am. Chem. Soc., 2000, 122, 10063–10072 CrossRef CAS.
- M. L. Hoppe, R. M. Laine, J. Kampf, M. S. Gordon and L. W. Burggraf, Angew. Chem., Int. Ed. Engl., 1993, 32, 287–289 CrossRef.
- R. M. Laine, K. Y. Blohowiak, T. R. Robinson, M. L. Hoppe, P. Nardi, J. Kampf and J. Uhm, Nature, 1991, 353, 642–644 CrossRef CAS.
- D. L. Bailey, A. Snyder and F. M. O'Connor, US Pat., 2,881,198, 1959.
- E. Suzuki, M. Akiyama and Y. Ono, J. Chem. Soc., Chem. Commun., 1992, 136–137 RSC.
- C. L. Frye, G. A. Vincent and W. A. Finzel, J. Am. Chem. Soc., 1971, 93, 6805–6811 CrossRef CAS.
- B. B. Williams, J. L. Gidley and R. S. Schechter, Acidizing Fundamentals, in SPE Monograph Vol. 6, SPE, New York, 1979 Search PubMed.
- P. M. Collins and R. J. Ferrier, Monosaccharides, Wiley, Chichester, 1995 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |