An improved dual channel PERCA instrument for atmospheric measurements of peroxy radicals
Timothy J.
Green
a,
Claire E.
Reeves
a,
Zoe L.
Fleming†
b,
Neil
Brough
a,
Andrew R.
Rickard‡
b,
Brian J.
Bandy
a,
Paul S.
Monks
b and
Stuart A.
Penkett
a
aSchool of Environmental Science, University of East Anglia, University Plain, Norwich, UK NR4 7TJ
bDepartment of Chemistry, University of Leicester, Leicester, UK LE1 7RH
First published on 21st April 2006
Abstract
This paper describes a new dual-channel PEroxy RadiCal Amplification (PERCA) instrument, which has been designed to improve the time resolution and signal to noise and to reduce the interference caused by variations in ambient ozone concentrations. The instrument was run at the Weybourne Atmospheric Observatory (WAO), North Norfolk, during WAOWEX (Weybourne Atmospheric Observatory Winter Experiment) in January/February 2002 and INSPECTRO (Influence of clouds on the spectral actinic flux in the lower troposphere) in September 2002. The performance of the instrument is assessed and compared to that of a single channel instrument. In particular, it is shown how the precision is greatly improved in fluctuating background ozone conditions. In addition the improved time response of the instrument allows changes in peroxy radical concentrations to be related to rapid changes in nitric oxide concentrations and the ozone photolysis frequency, j(O1D).
Introduction
Peroxy radicals (e.g., HO2 and RO2, where RO2 is a generic organic peroxy radical) are key reactive intermediates/chain propagators in ozone (O3) photochemistry.1,2 They are formed in the oxidation of many natural and anthropogenic trace species (e.g., methane (CH4), carbon monoxide (CO), non-methane hydrocarbons). Reaction of the peroxy radicals with nitric oxide (NO) leads to O3 formation, whilst the hydroperoxy radical (HO2) can react with O3 to destroy it. The self- and cross-reactions of the peroxy radicals forming peroxides can constitute a major sink for HOx (HO2 + OH (hydroxy radical) (e.g., ref. 3,4). Therefore the gathering and interpretation of peroxy radical data under a wide range of atmospheric conditions remains a key issue for tropospheric chemistry.Considerable progress has been made in the measurement of atmospheric radical species in situ at ground level (e.g., ref. 5–16) and on airborne platforms (e.g., ref. 17–22). The groups at the University of East Anglia and the University of Leicester have made extensive peroxy radical measurements using the PEroxy Radical Chemical Amplification technique (PERCA) in relatively clean atmospheres of the remote marine boundary layer (MBL),11,13,23–28 and in the free troposphere.12,22,29 As the level of pollution (particularly NOx and ozone) increases there has been a tendency for the atmospherically induced noise in the PERCA signal to increase, such that the data have to be averaged. Thus it has not been possible to examine high frequency atmospheric variability in the peroxy radicals in polluted environments. Also, a lack of high frequency data limits the usefulness of aircraft measurement since a 1 min average can translate to a distance of 6 km over which the air is sampled.22
We report here the development of a peroxy radical instrument, based on the PERCA technique, which has a precision of the order of ca. 1 parts per trillion by volume (pptv) on a 1 min time-base, even in conditions of moderate pollution, thus representing a major improvement in our measuring ability in near-detection limit conditions. The accuracy is of the order of 42%, similar to previous PERCA instruments. Data are presented showing that peroxy radical concentrations are perturbed dramatically over the space of several minutes. We present the results obtained from running the improved instrument in two field campaigns at Weybourne, Norfolk, England (WAOWEX (Weyborne Atmospheric Observatory Winter Experiment) in January/February 2002 and INSPECTRO (Influence of clouds on the spectral actinic flux in the lower troposphere) in September 2002.
Experimental
Section 1: PERCA technique
The PERCA technique was pioneered by Cantrell, Stedman and Wendel.30–32 It uses the catalytic conversion of ambient mixing ratios of reactive radicals to nitrogen dioxide (NO2) and carbon dioxide (CO2) by doping the sampled gas flow with high concentrations of intimately mixed NO and CO. Amplification of the peroxy radicals is achieved when the OH produced in the NO to NO2 conversion reaction reacts with the CO to form HO2. e.g.,| HO2 + NO → OH + NO2 | (1) |
| OH + CO → H + CO2 | (2) |
| H + O2 + M → HO2 + M | (3) |
| Overall: NO + CO + O2 → NO2 + CO2 | (4) |
The chemical complexity of the CL chemistry and the dependence of the CL on water vapour are the principle drawbacks to the technique. Salisbury et al.11 showed that the CL of the UEA-Leicester PERCA II instrument fell approximately linearly with increasing specific humidity and thus all subsequent data produced by these instruments have been corrected for this effect. The gas-phase reactions believed to occur within a PERCA inlet are well known and characterised,33 since they are all reactions of atmospheric importance and have thus been thoroughly studied in the laboratory for some time. However, the geometry and material of the inlets used affect the wall-loss rates of the highly reactive radical species H, OH and HO2 participating in the inlet chemistry. Loss of radical fragments to the wall in amplification mode is the main limiting factor (excepting the inlet reagent mixing ratios) in determining the CL. Pyrex and PFA are commonly used inlet materials, and the inlet geometry is one which gives a relatively high surface to volume ratio. Although this results in a lower CL than could be achieved if wall-loss were minimized, the small volume allows a rapid switch between amplification and termination modes and ensures a dominant and near constant termination process. If the termination mode signal is not well defined, the amount of NO2 produced in the amplification chemistry cannot be precisely ascertained.
The sampled radical population, once amplified, typically contributes 3–9 ppbv NO2, whereas sampled ozone, which is titrated to NO2 under the high NO concentrations in the inlet can contribute up to 100 ppbv. If the residence time in the inlet is of the order of 1 s, thermal decomposition of NO2 reservoir species, such as PAN, is small and can be neglected. It can be seen that changes in ozone concentration, if they occur between amplification and termination periods, can quickly swamp the amount of NO2 produced by the amplification chemistry on the radicals. Cantrell et al.34 recognized this fact and implemented a dual channel PERCA where one channel was locked in amplification mode and the other channel was locked in termination mode: the data from the two detectors were combined so that a continuous measure of ozone fluctuations can be recorded. To account for drifts in detector sensitivity, the two detectors had to be periodically swapped. The large internal volume of the inlets of this original design would have made time-matching of the two signals extremely difficult. Our system is superior in that it consists of two completely independent inlets of low volume, which are also calibrated individually.
Our calibration source7 also differs from that employed by Cantrell et al.34 The instrument is normally calibrated on a twice weekly cycle in the field. Calibration cycles (NO2 and methyl iodide (CH3I)) typically take up to 3 h. An NO2 calibration is done every day lasting 0.5 h. For a typical campaign (5 weeks), the overall data accumulation, including unforeseen instrument downtime, is of the order of >96%. Zero-air at three different flow rates was mixed with a stream of NO2 in 50 standard cubic centimetres per minute (sccm) of N2. The permeation rate of the NO2 was around 400 ng min−1, creating NO2 concentrations of between 0 and 120 ppbv NO2 which are used to calculate the sensitivity of the detectors (in volts) to concentration of NO2.
In both single and dual-channel systems, a simple subtraction of the termination from the amplification signal provides a signal due solely to amplified NO2 from peroxy radicals at all times. With a single channel PERCA, background O3 fluctuations often necessitate the use of signal averaging to yield satisfactory precision. Typically, about 1 min of amplification mode data will be averaged to produce one point discriminated from the interpolated average of two 1 min periods of termination mode. The best time resolution that can be achieved is thus 2 min, and in practice it is necessary to average the data to 10 min and often 30 min periods to reduce the scatter in the data to acceptable levels (standard deviations of ∼1 pptv). By using two identical inlets run out of phase with one amplifying while the other is terminating and vice versa, a near real-time signal from the radicals can be recorded continuously.
Section 2: Instrument description
The instrument used in this study is a 4th generation PERCA developed jointly by the University of East Anglia and the University of Leicester and benefits from a number of improvements which have been made over the period 1993 to 2003. It utilizes a number of innovative features which will be described here, but the basic methodology is very similar to the single channel instruments that have preceded it.7,12,22 The instrument gas and liquid flows are shown schematically in Fig. 1. | ||
| Fig. 1 Schematic of the PERCA-4 instrument showing how the two inlets are connected to and share the other units. The square boxes containing letters indicate gas lines. Those in the legend indicate addition and removal points of the gases. Those not in the legend (D, E and G) represent points that are simply connected together. | ||
PERCA-4 represents a major improvement over single channel instruments using the same chemical amplification mechanisms and inlet geometries in that it maintains a precision of ∼1 pptv for 1 min data over nearly all air masses encountered. The estimated overall accuracy of 42%35 however, is similar to that optimally achieved with a single channel instrument, since the errors are in the radical calibration and measurement of NO2. The uncertainties in the chain length (photolysis of CH3I (15%), CH3I (5%) and zero-air flow rates (10%), CH3I permeation tube leak rate (5%), photolysis cell volume (5%)), NO2 detection (thermal stability of luminal (20%)) and humidity correction (25%) are all accounted for in the overall accuracy estimate. For the dual channel instrument the uncertainties in the flow rates and NO2 detection need to be considered twice.
The new instrument is also a major improvement over the only other previously published example of a dual-channel PERCA.34 This is because of the different purposes for which the two systems were developed. The DICHAMP (Dual Inlet Chemical AMPlifier) of Cantrell et al.34 was constructed to counteract the severe problems encountered using their single channel instrument which had used inlets with a small surface to volume ratio to minimise wall losses within the inlet and maximise the amplification of CL. Use of such a large volume inlet arrangement precludes amplification to background switching times of less than 3 min. Over a 3 min period background ozone levels can often change by about 1–3 ppbv, even in relatively homogeneous air masses. The maximum amount of NO2 typically produced by the CL chemistry from a noon-time peroxy radical population is very similar to that produced from sudden increases in ozone. To compound the problem, the instrument of Cantrell et al. relied on the ozone background being determined by the CO being removed from the sampled flow altogether (and switched to the other inlet). Thus in dual-channel mode, the background for the channel not in amplification mode must be determined solely from the other channel making it imperative that the sensitivity to NO2 of each detector be known with a high degree of accuracy. Unfortunately, luminol-based LMA-3 instruments are prone to instrumental drift, no matter how carefully the luminol is supplied to them.
The PERCA methodology applied here has followed a different rationale. Low volume inlets with a small residence time which allow rapid switching between amplification and termination modes have been used (typically every 30 s or 1 min, depending on air-mass composition). To determine the background the CO flow is added through a second injection point about 1 s downstream from the NO injection to allow sufficient time for the sampled radicals to form nitrate products that are not amplified by the CO. Also the termination process is dominated by wall loss which is nearly constant.
The dual channel PERCA-4 instrument was based on combining two completely independent inlet systems taking into account the phase of each signal. As amplification and background signals are still obtained with each inlet, the data can be worked up from both inlets individually, and then combined together to yield a dual-channel signal (see Fig. 2). Additionally, any periods of sensitivity drift can be easily flagged.
 | ||
| Fig. 2 Signals from the 2 channels of the PERCA instrument and averages used to calculate the dual channel signal. The lower and middle panels are for Channel 1 and Channel 2 respectively. In these two panels the thin black traces are the raw signals (RS) with the axes scaled to account for sensitivity (Sens); amplification (A) and background (B) averages are shown as the upper and lower grey lines, respectively; and the thick black lines represent the averages of the amplification and the background interpolated averages (M). Dual-channel data are shown as the thin black trace in the top panel. | ||
A further improvement is that PERCA-4 utilised a novel method of supplying luminol solution to the two LMA-3 detectors. The usual method of supply relies on a peristaltic pump which can introduce cyclical noise to the raw LMA-3 signal. The system begins to increase in complexity when using more than one detector, and it was felt that pressurised displacement of the luminol, with regulation of the flow by needle valves, was a simpler, more efficient method. N2 (25 psig) is added to a luminol solution to deliver it under pressure via two separate needle valves to the detector (see Fig. 1). Luminol removal is regulated under suction by two needle valves. The instrument sampling pump provides the suction which is reduced by a flow restrictor. The new system designed for PERCA-4 produces much smoother flows and has proved extremely reliable. Data rejection is kept to a minimum (typically 1–3 min in any hour) with regular maintenance of the LMA-3 cells.
Results and discussion
Section 1: Instrument performance
Fig. 2 illustrates how the raw NO2 signals from each channel are manipulated to produce the dual channel data. The thin black traces are the raw signals from each channel (RS1 and RS2) with the axes scaled to account for sensitivity differences between them (Sens1 and Sens2) found from the absolute NO2 calibrations. Amplification and background averages are shown as upper (A1 and A2) and lower (B1 and B2) grey lines. The thick black lines represent the averages of the amplification and the background interpolated averages (M1 and M2). Data sets for each single channel are produced by first removing the background distortion from each raw signal (i.e., RS1 − M1 and RS2 − M2). This produces data centred on zero and removes bias introduced into the data from changing ozone concentrations. The amplification averages and background averages are then extracted from these centred waves to produce the amplified NO2 for each channel ([NO2]amp1 and [NO2]amp2). Dividing by each sensitivity and each CL then gives the sampled peroxy radical concentration: {[NO2]amp1/CL1}/Sens1 and {[NO2]amp2/CL2}/Sens2. Dual-channel data are extracted from the signals as {[NO2]amp1/CL1}/Sens1 − {[NO2]amp2/CL2}/Sens2 and is shown in Fig. 2 as the thin black trace in the upper panel. The regions of negative data show ambient radical data produced when the second channel is amplifying, the positive periods show ambient data from when the first channel is amplifying. To extract near continuous radical data, one has only to discard the periods when the channels are switched (approximately 6 s of data every 1 min) and invert the negative period. The channels are switched to minimise systematic errors.Fig. 3 shows PERCA data from the INSPECTRO deployment at Weybourne on 27th September 2002. The signals from channels 1 and 2 show a very strong correlation with ozone due to its titration to NO2 in the inlet. With a single channel this changing signal due to ozone over the course of an amplification and background cycle masks the changes due to the amplification of the radicals. Note how the relative sensitivities of the two channels vary throughout the 9 h shown. Fig. 3 also shows concentrations of peroxy radicals as calculated using a single channel and a dual channel approach. The precision of the dual channel data is clearly much improved.
 | ||
| Fig. 3 The raw signals from the two channels (thin black and grey lines) and ozone concentration (thick black line) for 27th September 2002 at Weybourne. Peroxy radical mixing ratios: worked up from the individual channels (grey symbols); worked up using the dual channel technique (black symbols). | ||
Table 1 shows a comparison of the data resulting from each channel individually with the data produced using both channels. The data are from 04:30 to 06:30 GMT on the morning of the 27th September 2002, a period characterised by stagnant, low wind speed conditions (see also Fig. 3). Over the period 05:30 to 06:30 GMT a great deal of structure was observed in the background LMA-3 signal, representing fluctuations in ambient ozone of about 7 ppbv over 10–20 s periods. While such data are not particularly representative of any atmospheric regime, it is akin to the kind of ozone variability that can be encountered when a PERCA is deployed on an aircraft22 and it demonstrates the superior performance of the new dual channel PERCA. Table 1 shows quantitatively that the precision of the dual channel data is of the order of 0.5 pptv (calculated as the standard deviation of 5 data points over a 5 min period), even when conditions are highly unfavourable in terms of background ozone fluctuations. It can be seen that for the latter period the single channel data, under such extreme conditions, are completely unreliable. Even averaging the data to 30 min yields no improvement. The dual channel instrument’s superior precision is maintained to a great extent over a wide variety of air masses. This is not true of the single channel instrument which can yield data with excellent precision in ideal circumstances,22,23,25 but in sub-optimal conditions requires extensive data averaging to pull data out from the instrumental noise. This variability in precision has been a serious drawback to the PERCA technique when used as a single channel instrument. Worse still, there is no simple way to tell when data variability is as a result of background variation due to ambient ozone fluctuations, or to the very real perturbations that occur as a result of chemical and radiative forcing.
| Mixing ratio (standard deviation) pptv (1σ) | |||
|---|---|---|---|
| Channel 1 | Channel 2 | Dual Channel | |
| Data during 04:30–05:30 | 1.54 (1.56) | 1.72 (1.00) | 1.48 (0.40) |
| Data during 05:30–06:30 | −5.78 (14.26) | 7.04 (13.03) | 0.90 (0.51) |
Section 2: Field measurements
PERCA-4 testing at Weybourne during the summer of 2001 produced the first clear evidence that the dual channel instrument represented a major step forward over the single channel methodology. Night-time radical concentrations could be easily discriminated on a 1 min time base. The instrument deployment at Weybourne in February 2002 during WAOWEX produced clear evidence that night-time radical levels are higher than daytime levels during winter.35 For the summer 2002 campaigns at Weybourne the instrument had reached a higher level of precision and produced peroxy radical data with sub-pptv precision on a 1 min time base with better than 80% campaign coverage.Fig. 4 shows a time series of 1 min peroxy radical data from the INSPECTRO campaign at WAO from late 16 September to 2 October 2002. It can be seen that for most of the campaign the daytime peroxy radical levels respond to changing solar radiation, as predicted by basic photochemical theory and observed previously.25 This is particularly clear on the 16th, 17th, 18th, 19th, 21st, 22nd, 23rd and 24th of September. Large concentrations of peroxy radicals (up to 40 pptv, off the scale in Fig. 4) are observed at night and towards the end of the campaign, associated with increased concentrations of NOx (Fig. 5): these exceed those observed earlier during the daytime and are almost certainly associated with nitrate chemistry.36
 | ||
| Fig. 4 Peroxy radical concentrations (solid black dots) and j(O(1D)) photolysis frequencies (solid grey dots) for the INSPECTRO campaign at Weybourne during 2002. | ||
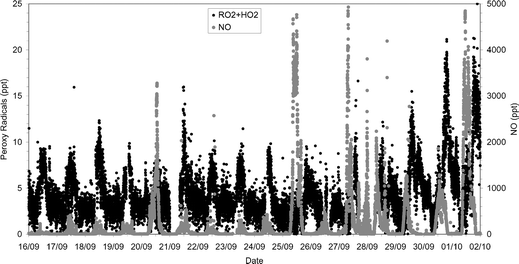 | ||
| Fig. 5 Peroxy radical concentrations (solid black dots) and NO concentrations (solid grey dots) for the INSPECTRO campaign at Weybourne during 2002. | ||
Fig. 5 shows that the daytime peroxy radical signal is also clearly affected by the NO levels. For example the peroxy radicals on the 20th and 25th September do not exhibit the daytime increase associated with the increase in j(O1D) (Fig. 4), whilst NO concentrations are enhanced (several ppbv).
The WAO site is frequently impacted by fresh local NOx emissions, from shipping and road vehicles etc., depending on the wind sector. Such fresh emissions are typically characterised by short transitory plumes of 2–3 min duration and the fast time resolution of the UEA/Leicester PERCA-4 instrument allows the peroxy radical population to be measured at discrete points during these episodes. This is clearly demonstrated in Fig. 6, where it can be seen that over the course of a few hours on the 18th September 2002 the peroxy radicals track j(O1D). However, there are occasionally short periods when NO concentrations are elevated, which are associated with decreases in the peroxy radical concentrations. NO reacts with HO2 and RO2 to form NO2 and either OH or RO (alkoxy radical), thus leading to these observed anti-correlations between NO and the peroxy radical concentrations. The details of the relationships between peroxy radical concentrations and NO and j(O1D) are the subject of a subsequent paper by this group of authors and of Fleming et al.,35 but it should be noted here that the improved performance in the instrument described in this paper has reduced the noise in these relationships and allowed them to be examined over shorter time intervals for the first time.
 | ||
| Fig. 6 Peroxy radical concentrations (solid black circles), j(O(1D)) photolysis frequencies (solid grey circles) and NO concentrations (open circles) for the INSPECTRO campaign at Weybourne during 18th September 2002. | ||
Conclusion
This paper describes a new dual-channel PERCA instrument, which is based on combining two completely independent inlet systems. The data from each channel are worked up individually and then combined to yield a dual channel signal. With the background and amplification modes out of phase between the two channels, a measure of the peroxy radical concentration can be made almost continuously as opposed to once per amplification cycle. Furthermore the interference from O3 is removed when the signals from the two channels are combined leading to much improved precision (0.5 pptv). The improved time response and precision of the instrument has allowed changes in peroxy radical concentrations to be related to rapid changes in NO concentrations and j(O1D).As interest in atmospheric composition becomes focused on urban regions, the timescales for chemical processes to occur typically tend to approach those of the transport processes. To access the nature of the chemistry occurring, it is imperative that fast instrumentation be employed. PERCA-4 represents a major step forward in the state-of-the-art of PERCA measurements in general. In particular, it is now possible to measure peroxy radicals with high precision in conditions in which it would have been impossible to extract such data with a single channel instrument.
Acknowledgements
This work was funded by the NERC Core Strategic Measurements in Atmospheric Science (COSMAS) and the EU Framework 5 project INSPECTRO. Thanks also to UEA mechanical and electrical workshops, especially Mr Brendan O’Brien and Mr Graham Evans and to Dr Georgina Sturrock and Dr Graham Mills for much helpful assistance during the preliminary testing and INSPECTRO fieldwork.References
- J. A. Logan, M. J. Prather, S. C. Wofsy and M. B. McElroy, J. Geophys. Res., C: Oceans Atmos., 1981, 86, 7210 Search PubMed.
- P. S. Monks, Chem. Soc. Rev., 2005, 34, 376 RSC.
- G. Chen, D. Davis, J. Crawford, B. Heikes, D. O’Sullivan, M. Lee, F. Eisele, L. Mauldin, D. Tanner, J. Collins, J. Barrick, B. Anderson, D. Blake, J. Bradshaw, S. Sandholm, M. Carroll, G. Albercook and A. Clarke, J. Atmos. Chem., 2001, 38, 317 CrossRef CAS.
- C. E. Reeves and S. A. Penkett, Chem. Rev., 2003, 103, 5199 CrossRef CAS.
- J. Burkert, M. D. Andres-Hernandez, L. Reichert, J. Meyer-Arnek, B. Doddridge, R. R. Dickerson, J. Muhle, A. Zahn, T. Carsey and J. P. Burrows, J. Geophys. Res., [Atmos.], 2003, 108.
- C. A. Cantrell, R. E. Shetter, T. M. Gilpin, J. G. Calvert, F. L. Eisele and D. J. Tanner, J. Geophys. Res., [Atmos.], 1996, 101, 14653 CrossRef CAS.
- K. C. Clemitshaw, L. J. Carpenter, S. A. Penkett and M. E. Jenkin, J. Geophys. Res., [Atmos.], 1997, 102, 25405 CAS.
- D. J. Creasey, P. A. HalfordMaw, D. E. Heard, M. J. Pilling and B. J. Whitaker, J. Chem. Soc., Faraday Trans., 1997, 93, 2907 RSC.
- D. J. Tanner, A. Jefferson and F. L. Eisele, J. Geophys. Res., [Atmos.], 1997, 102, 6415 CrossRef CAS.
- C. M. Mihele and D. R. Hastie, J. Atmos. Oceanic Technol., 2000, 17, 788 Search PubMed.
- G. Salisbury, P. S. Monks, S. Bauguitte, B. J. Bandy and S. A. Penkett, J. Atmos. Chem., 2002, 41, 163 CrossRef CAS.
- P. Zanis, P. S. Monks, E. Schuepbach and S. A. Penkett, J. Geophys. Res., [Atmos.], 1999, 104, 26913 CrossRef CAS.
- S. A. Penkett, K. C. Clemitshaw, N. H. Savage, R. A. Burgess, L. M. Cardenas, L. J. Carpenter, G. G. McFadyen and J. N. Cape, J. Atmos. Chem., 1999, 33, 111 CrossRef CAS.
- D. E. Heard and M. J. Pilling, Chem. Rev., 2003, 103, 5163 CrossRef CAS.
- M. D. A. Hernandez, J. Burkert, L. Reichert, D. Stobener, J. Meyer-Arnek, J. P. Burrows, R. R. Dickerson and B. G. Doddridge, J. Geophys. Res., [Atmos.], 2001, 106, 20833 CrossRef CAS.
- J. Burkert, M. D. Andres-Hernandez, D. Stobener, J. P. Burrows, M. Weissenmayer and A. Kraus, J. Geophys. Res., [Atmos.], 2001, 106, 5457 CrossRef CAS.
- W. H. Brune, I. C. Faloona, D. Tan, A. J. Weinheimer, T. Campos, B. A. Ridley, S. A. Vay, J. E. Collins, G. W. Sachse, L. Jaegle and D. J. Jacob, Geophys. Res. Lett., 1998, 25, 1701 CrossRef CAS.
- T. Reiner, M. Hanke, F. Arnold, H. Ziereis, H. Schlager and W. Junkerman, J. Geophys. Res., [Atmos.], 1999, 104, 18647 CrossRef CAS.
- I. Faloona, D. Tan, W. H. Brune, L. Jaegle, D. J. Jacob, Y. Kondo, M. Koike, R. Chatfield, R. Pueschel, G. Ferry, G. Sachse, S. Vay, B. Anderson, J. Hannon and H. Fuelberg, J. Geophys. Res., [Atmos.], 2000, 105, 3771 CrossRef CAS.
- L. Jaegle, D. J. Jacob, W. H. Brune, I. Faloona, D. Tan, B. G. Heikes, Y. Kondo, G. W. Sachse, B. Anderson, G. L. Gregory, H. B. Singh, R. Pueschel, G. Ferry, D. R. Blake and R. E. Shetter, J. Geophys. Res., [Atmos.], 2000, 105, 3877 CrossRef CAS.
- C. A. Cantrell, G. D. Edwards, S. Stephens, R. L. Mauldin, M. A. Zondlo, E. Kosciuch, F. L. Eisele, R. E. Shetter, B. L. Lefer, S. Hall, F. Flocke, A. Weinheimer, A. Fried, E. Apel, Y. Kondo, D. R. Blake, N. J. Blake, I. J. Simpson, A. R. Bandy, D. C. Thornton, B. G. Heikes, H. B. Singh, W. H. Brune, H. Harder, M. Martinez, D. J. Jacob, M. A. Avery, J. D. Barrick, G. W. Sachse, J. R. Olson, J. H. Crawford and A. D. Clarke, J. Geophys. Res., [Atmos.], 2003, 108, 8797 CrossRef.
- T. J. Green, C. E. Reeves, N. Brough, G. D. Edwards, P. S. Monks and S. A. Penkett, J. Environ. Monit., 2003, 5, 75 RSC.
- P. S. Monks, L. J. Carpenter, S. A. Penkett and G. P. Ayers, Geophys. Res. Lett., 1996, 23, 535 CrossRef CAS.
- L. J. Carpenter, P. S. Monks, B. J. Bandy, S. A. Penkett, I. E. Galbally and C. P. Meyer, J. Geophys. Res., [Atmos.], 1997, 102, 25417 CrossRef CAS.
- S. A. Penkett, P. S. Monks, L. J. Carpenter, K. C. Clemitshaw, G. P. Ayers, R. W. Gillett, I. E. Galbally and C. P. Meyer, J. Geophys. Res., [Atmos.], 1997, 102, 12805 CrossRef CAS.
- P. S. Monks, L. J. Carpenter, S. A. Penkett, G. P. Ayers, R. W. Gillett, I. E. Galbally and C. P. Meyer, Atmos. Environ., 1998, 32, 3647 CrossRef CAS.
- P. S. Monks, G. Salisbury, G. Holland, S. A. Penkett and G. P. Ayers, Atmos. Environ., 2000, 34, 2547 CrossRef CAS.
- G. Salisbury, A. R. Rickard, P. S. Monks, B. J. Allan, S. Bauguitte, S. A. Penkett, N. Carslaw, A. C. Lewis, D. J. Creasey, D. E. Heard, P. J. Jacobs and J. D. Lee, J. Geophys. Res., [Atmos.], 2001, 106, 12669 CrossRef CAS.
- P. Zanis, P. S. Monks, T. J. Green, E. Schuepbach, L. J. Carpenter, G. P. Mills, A. R. Rickard, N. Brough and S. A. Penkett, Geophys. Res. Lett., 2003, 30, 1497 CrossRef.
- C. A. Cantrell, D. H. Stedman and G. J. Wendel, Anal. Chem., 1984, 56, 1496 CrossRef CAS.
- C. A. Cantrell, R. E. Shetter, A. H. McDaniel and J. G. Calvert, Adv. Chem. Ser., 1993, 291 Search PubMed.
- C. A. Cantrell, R. E. Shetter, J. A. Lind, A. H. McDaniel, J. G. Calvert, D. D. Parrish, F. C. Fehsenfeld, M. P. Buhr and M. Trainer, J. Geophys. Res., [Atmos.], 1993, 98, 2897 CrossRef CAS.
- L. Reichert, M. D. A. Hernandez, D. Stobener, J. Burkert and J. P. Burrows, J. Geophys. Res., [Atmos.], 2003, 108 Search PubMed.
- C. A. Cantrell, R. E. Shetter and J. G. Calvert, Anal. Chem., 1996, 68, 4194 CrossRef CAS.
- L. Fleming, P. S. Monks, A. R. Rickard, B. J. Bandy, N. Brough, T. J. Green, C. E. Reeves and S. A. Penkett, Atmos. Chem. Phy., 2006, submitted Search PubMed.
- N. Carslaw, L. J. Carpenter, J. M. C. Plane, B. J. Allan, R. A. Burgess, K. C. Clemitshaw, H. Coe and S. A. Penkett, J. Geophys. Res., [Atmos.], 1997, 102, 18917 CAS.
Footnotes |
| † Present address: Imperial College, London, UK SW7 2AZ. |
| ‡ Present address: Department of Chemistry, University of Leeds, Leeds, UK LS2 9JT. |
| This journal is © The Royal Society of Chemistry 2006 |