Proton sponge phosphines: electrospray-active ligands†‡
Nicola J.
Farrer
ab,
Robert
McDonald
c and
J. Scott
McIndoe
*b
aDepartment of Chemistry, The University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW
bDepartment of Chemistry, University of Victoria, P. O. Box 3065, Victoria, BC, Canada V8W 3V6. E-mail: mcindoe@uvic.ca
cX-Ray Crystallography Laboratory, Department of Chemistry, University of Alberta, Edmonton, AB, Canada T6G 2G2
First published on 24th August 2006
Abstract
Attachment of a proton sponge to a phosphine ligand renders neutral complexes of the ligand highly amenable to analysis by electrospray ionisation mass spectrometry (ESI-MS). The ligand 1,8-bis(dimethylamino)naphthyldiphenylphosphine (3) is extremely efficient and highly selective in forming exclusively [M + H]+ ions, which may be detected at very low concentration. Ionisation efficiency of 3 in the presence of H+ approached 100%. The bis-substituted ligand bis{1,8-bis(dimethylamino)naphthyl}phenylphosphine (4) was also prepared and characterised, as were Fe(CO)4- (5c), Mn(η5-C5H4Me)(CO)2- (6) and W(CO)5- (7) complexes of 3. Compounds 3, 3·HBr·EtOH, 4 and 5c were all structurally characterised.
Introduction
Electrospray ionisation mass spectrometry (ESI-MS) is an increasingly common tool for the direct analysis of catalytically active species.1 The majority of analyses of this type have employed catalysts which themselves carry a charge, i.e. are cations or anions. These species are transported directly into the gas phase with very high efficiency and hence are ideally suited to analysis by ESI-MS. High m/z relative to ions derived from solvent and substrates also assists the efficient detection of catalysts based on organometallic complexes. However, a sizable proportion of organometallic catalysts are neutral complexes, and hence rely on one of the various ionisation pathways that generate detectable ions.2 Numerous such pathways exist, most well-known being protonation to form [M + H]+ ions, and related ions such as [M + Na]+, [M + K]+ or [M + NH4]+ (depending on which ions are added or the presence of adventitious ions). Loss of halide from metal halide complexes, LnMXm, to form [LnMXm−1]+ ions is another common ionisation pathway.3 Complexes with acidic protons may undergo loss of H+ to generate [M − H]− ions.4 Particularly electron-rich complexes may undergo electrochemical oxidation to generate [M]+ radical cations, most notably those based on ferrocene.5 In certain cases, addition of an ionisation agent can result in highly efficient production of charged derivatives, such as the addition of alkoxy ions, RO−, to metal carbonyl complexes to generate [M + OR]− ions,6 or of Ag+ to metal–metal bonded species to form [M + Ag]+ ions.7Henderson and co-workers introduced the idea of using the commercially available phosphines P(p-C6H4OMe)3 and P(p-C6H4NMe2)3 in place of triphenylphosphine as “electrospray-friendly” ligands.8 This approach allowed the observance of [M + H]+ and [M + Na]+ ions for most of the complexes of these ligands; similarly, cobalt carbonyl clusters with {µ3-Si(p-C6H4OMe)} and {µ3-Si(p-C6H4NMe2)} ligands provided ESI mass spectra by protonation.9 However, aryl amines and aryl ethers are not especially basic and the efficiency with which these sites are protonated is low. As such, these ligands are not well suited to the demands of direct ESI-MS analysis of complexes under non-ideal circumstances, e.g. in a catalytic reaction, a complex mixture, or in the presence of other compounds that provide very strong spectra, such as a complex dissolved in an ionic liquid. The ideal functional group would have high basicity, be selective for a single ion, and be non-nucleophilic (to minimise interference in reactions). The group should also not greatly affect the electronic properties of the electrospray-inactive ligand it replaces. Aromatic proton sponges fit these requirements rather well.10 We selected 1,8-bis(dimethylamino)naphthalene (1), introduced by Alder11 and sold by Aldrich as “Proton Sponge®”; it is not only the best-known compound of its type but is inexpensive and straightforward to derivatise. Its pKaH of 12.1 (in water) is a million-fold higher than most aromatic amines, due to relief of steric strain in the neutral base upon protonation.121 is non-nucleophilic; it may be recovered unchanged after four days at reflux with ethyl iodide in acetonitrile,11 and it has been utilised in organic chemistry as a non-nucleophilic base.13
Some examples of reactivity of 1 apart from routine protonation include, somewhat surprisingly, its role as a hydride donor in its reaction with mer-RhCl3(dmso)3 or [RuCl(dppb)]2(µ-Cl)3,14 with fluoroalkyl complexes of iridium,15 or with B(C6F5)316 to form the 1,1,3-trimethyl-2,3-dihydroperimidinium cation (TMP+). There is also a single example of a metal complex coordinating (directly) to 1 (via the amino groups); the reaction with Pd(hfac)2 immediately generates a poorly-characterised charge-transfer product, which after standing for a week forms the cationic complex [Pd(hfac)(1)]+.17 The hfac ligand may be substituted for β-diketones and one of these complexes was structurally characterised; coordination causes severe distortion of the proton sponge, the N⋯N distance opening to 2.94 Å from 2.51 Å. The proton sponge ligand is easily displaced in this complex, even by water. 1 can also act as a weak carbon nucleophile, but only in the presence of exceptionally reactive electrophiles.18
An additional advantage of a proton sponge-substituted ligand is that the proton sponge confers pH-dependent solubility properties: in neutral or basic media, the sponge is soluble in organic solvents; in acidic media, it carries a charge and is soluble in polar solvents, including water (depending on the counterion involved). These characteristics are passed on to the ligand and hence also to metal complexes of the ligand.
Herein we report the syntheses of {1,8-bis(dimethylamino)naphthalen-2-yl}diphenylphosphine (3) and bis{1,8-bis(dimethylamino)naphthalen-2-yl}phenylphosphine (4), metal complexes of ligand 3 and a comparison of its performance as an ESI-MS handle in organometallic complexes with the PPh3 ligand and the “electrospray-friendly” ligand P(p-C6H4OMe)3.
Experimental
Materials
Dry solvents were obtained by distillation or from a solvent purification system. Unless indicated, solvents were HPLC grade. Reagents were purchased from Strem and Aldrich and used without further purification except for Proton Sponge® (1), which was recrystallised from hot MeOH before use. 5a was prepared using a published method. Unless indicated otherwise, reactions were performed under nitrogen using standard Schlenk techniques. Electrospray ionisation mass spectra were collected using either a Micromass Quattro LC or a Micromass QTof micro instrument. Relative intensities (%) were determined by integration (using Origin 7.5®). Capillary voltage was set at 2900 V, source and desolvation gas temperatures were at 80 and 150 °C, respectively. Samples were infused via syringe pump at 5–10 µL min−1. NMR spectra were recorded on one of the following Bruker spectrometers: DPX-400, DRX-500, AV-500, AC-300 and AMX-360. Chemical shifts are quoted in δ (ppm) using internal references, where appropriate, of CDCl3 (1H δ 7.26 ppm), CD3CN (1H δ 1.94 ppm), acetone (1H δ 2.05 ppm) and external references of TMS (1H, 13C) and 85% aqueous H3PO4 (31P). All coupling constants (J) are given in Hz. Where required, assignments were determined through use of 1H{31P}, 1H-NOESY, 1H-COSY, 13C-DEPT, 1H–13C HSQC and 1H–13C HMBC NMR experiments. 13C assignments were consolidated by running 13C NMR experiments on both the AV-500 and AC-300 instruments and determining 31P–13C coupling constants by comparing the spectra obtained on the different instruments. Melting points were recorded on a Gallenkamp Melting Point Apparatus and are uncorrected. IR spectra were recorded using a solution cell in a Perkin-Elmer 1000 FTIR spectrometer. Microanalyses were carried out by the Analytical Laboratory in the Cambridge Chemical Laboratory.Preparations
The HBr salt 3·HBr was obtained as detailed above from the crude reaction mixture and crystallised from hot EtOH. 3·HBr crystallised with one molecule of EtOH in the unit cell, Mp (3·HBr) 189–191 °C. 31P{1H} NMR (202 MHz, CDCl3) δP (3·HBr): −17.7, δP (3(O)·HBr): 32.6. Anal. Calc. for C28H40BrN2O3P: (3·EtOH): C, 64.00; H, 6.52; N, 5.33; P, 5.89. Found C, 63.52; H, 6.46; N, 5.12; P, 5.88%.
3·HBF4 showed greater solubility in organic solvents (moderately soluble in MeCN, sparingly soluble in CDCl3) than 3·HBr and could be obtained by adding 48% HBF4 (aq) to an ethanolic suspension of crude 3 from the synthesis described earlier. The solution was heated to reflux under N2 and cooled, the white solid which precipitated on standing was isolated by suction filtration and recrystallised from hot EtOH to give 3·HBF4 as a white crystalline solid (14.8 g, 30.4 mmol, 74%), mp (3·HBF4) 236–238 °C. 1H NMR (500 MHz, CD3CN) δH (3·HBF4): 19.67 (1H, H+), 8.02 (dd, 1H, J 8, 1, H5), 7.96 (dd, 1H, J 1, J 8, H7), 7.93 (d, 1H, J 9, H4), 7.77 (dd, 1H, J 8, H6), 7.30–7.40 Hz (m, 11H: H3, 2Ph), 3.49 (dd, 6H, J 3, J 0.6, N(CH3)2 on C1), 3.20 (d, 6H, J 3, N(CH3)2 on C8). 13C{1H} NMR (126 MHz, CD3CN) δC (3·HBF4): 150.4 (d, J 24, C1), 144.9 (s, C8), 137.2 (s, C10), 136.4 (d, J 11, C2), 135.4 (d, J 28, C11), 134.9 (s, C13), 134.3 (d, J 19, C12), 130.8 (s, C5), 130.5 (s, C4), 130.4 (s, C14), 130.1 (d, J 7, C3), 129.3 (s, C6), 123.3 (s, C7), 121.7 (d, J 8, C9), 47.1 (s, CH3, N(CH3)2 on C8), 46.1 (d, CH3, J 17, N(CH3)2 on C1), 31P NMR (202 MHz, CD3CN) δP (3·HBF4): −17.3, δP (3(O)·HBF4): 32.9. ESI-MS (MeOH) m/z: 399.5 [3 + H]+
1H NMR (CD3CN, 500 MHz) δH (4·2HBF4): 19.32 (H+, 2H), 8.04 (dd, J 9, 1, 2H, H5), 7.96 (dd, J 7, 1, 2H, H7), 7.94 (d, J 9, 2H, H4), 7.75 (dd, J 8, 2H, H6), 7.42–7.46 (m, 1H, H14), 7.40 (ddd, J 8, 7, 2, 2H, H13), 7.28 (dd, 2H, J 9, H3), 7.22 (ddd, J 8, 8, 1, 2H, H12), 3.34 (d, J 1.4, 6H, N(CH3)2 on C1), 3.30 (d, J 1.5, 6H, N(CH3)2 on C1), 3.21 (d, J 3.4, 6H, N(CH3)2 on C8), 3.19 (d, J 3.4, 6H, N(CH3)2 on C8). 13C{1H} NMR (CD3CN, 500 MHz) δC (4·2HBF4): 149.9 (d, J 24, C1), 144.4 (C8), 137.5 (C10), 136.6 (d, J 6, C9), 135.0 (d, J 21, C12), 134.5 (C3), 133.4 (d, J 28, C11), 131.3 (C5), 131.2 (C14), 130.8 (C4), 130.6 (d, J 8, C13), 129.5 (C6), 123.5 (C7), 122.4 (d, J 9, C2), 47.4 (N(CH3)2 on C8), 47.3 (N(CH3)2 on C8), 45.7 (d, J 16, N(CH3)2 on C1), 45.0 (d, J 13, N(CH3)2 on C1), 31P{1H} NMR (202 MHz, CDCl3) δP (4·2HBF4): −26.4, ESI-MS (CH2Cl2) m/z: 535.2 [4 + H]+, 268.1 [4 + 2H]2+
Equimolar amounts of 1 (13.2 mg, 0.062 mmol) and 3·HBF4 (30.0 mg, 0.062 mmol) were dissolved together in dry, N2-saturated CD3CN (0.75 ml) and 1H NMR spectra recorded. Equimolar amounts of 1·HBF4 (7.8 mg, 0.026 mmol) and 3 (10.3 mg, 0.026 mmol) were dissolved together in dry, N2-saturated CD3CN (0.75 ml) and 1H NMR spectra were recorded. Both experiments was repeated. The average integral ratios for the coordinated H+ of the two protonated species over the four experiments were (CD3CN, 360 MHz) δH: 19.7 (3·H, ∫1), 18.7 (1·H, ∫0.93) (1 std. dev. = 0.02). The pKaH of 3 was determined by calculation using this ratio and the known pKaH of 1 in CD3CN, given as 18.18.20 Analysis of the equilibrium gave Ka2 = Ka1/Keq where Ka1 (known) = [1][H+]/[1·H+], Ka2 = [3][H+]/[3·H+] and Keq (known) = (∫3·H/∫1·H).2
The brown solution was filtered through cotton wool and the residue washed with dry THF (20 ml) to give a green-brown filtrate. The solvent was removed under vacuum, 20 ml of dry MeCN was added to the residue and the solution filtered to leave a cream precipitate (crude Fe(CO)3{P(C6H4OMe)3}2, see below for purification). The MeCN solution was washed repeatedly with hexane (6 × 10 ml) to remove Fe3(CO)12 and the solvent volume was reduced on a vacuum line. Crude yellow 5b precipitated from the cold minimal MeCN. Dissolving the precipitate in CH2Cl2 (5 ml) and filtering into ice-cold MeOH (10 ml) before leaving for 12 h in the fridge gave yellow crystals of 5b (0.27 g, 18%, 0.5 mmol), mp (5b) 151–153 °C.
1H NMR (500 MHz, CDCl3) δH (5b): 3.82 (s, 9H, 3CH3), 6.91 (dd, 6H, 3JH3–H2 8.8, 4JH3–P 1.8, H3), 7.38 (dd, 6H, 3JH2–H3 8.8, 3JH2–P 8.0, H2), 13C NMR (125 MHz, CDCl3) δC (5b): 55.5 (s, CH3), 114.3 (d, 3JC3–P 11.3, C3), 126.0 (d, 1JC1–P 55.3, C1), 134.8 (d, 3JC3–P 11.3, C2), 161.6 (d, 1JC4–P 2.5 C4), 213.9 (d, 1JOC–P 18.9, 4CO). 31P{1H} NMR: (202 MHz, CDCl3) δP (5b): 67.06 IR : ν(CO) (50 : 50 hexane–CH2Cl2) 2047 (sh), 1972, 1938 (s, br). ESI-MS (MeOH) 542 [5b + Na]+, ESI-MS (CH2Cl2–HBF4) 521.1 (87%) [5b + H+], 369.2 (59%) [PO(C6H4OMe)3 + H]+, 353.2 (100%) [P(C6H4OMe)3 + H]+.
The cream-coloured residue of Fe(CO)3{P(C6H4OMe)3}2 was dissolved in minimal CH2Cl2 and precipitated from the pale green solution by addition of hexane. Dissolving in CH2Cl2 (5 ml), filtering into ice-cold MeOH (10 ml) and leaving for 3 h at 4 °C gave Fe(CO)3{P(C6H4OMe)3}2 as flaky pale yellow crystals (13%, 0.313 g, 0.37 mmol). mp (Fe(CO)3{P(C6H4OMe)3}2) 217–218 °C. 31P{1H} NMR (146 MHz, CH2Cl2, d6-acetone lock) δP: 74.8. ESI-MS (CH2Cl2–HBF4) m/z: 845.3 (100%) [Fe(CO)3{P(C6H4OMe)3}2 + H]+, 353.2 (7%) [P(C6H4OMe)3 + H]+.
The monosubstituted product, 5c, was obtained as fine yellow crystals from CH2Cl2–hexane (0.36 g, 27%, 0.7 mmol), mp (5c) 190–191 °C (decomp.). 1H NMR (500 MHz, CDCl3) δH (5c): 7.60 (dd, 4H, 2J 10, 3J 8, 4H12), 7.55 (d, 1H, J 8, H5), 7.51 (d, 1H, J 8.5, H4), 7.43 (m, 7H, 4H13, 2H14 and H6), 7.35 (d, 1H, J 7, H7), 7.06 (dd, 1H, J 8.5, J 10, H3), 2.58 (6H, H8), 2.41 (6H, H1); 13C NMR (126 MHz, CDCl3) δC (5c): 214.6 (d, 2JC–P 19, 4CO), 154.2 (d, 2JC–P 7, C1), 153.4 (C8), 139.8 (C10), 136.6 (d, 1JC–P 47, C11), 134.1 (d, 2JC–P 10, 4C12), 133.3 (d, 1JC–P 58, C2), 130.4 (2C14), 130.1 (d, 2JC–P 10, C3), 129.5 (d, 3JC–P 6, C9), 128.4 (d, 2JC–P 10, 4C13), 127.6 (C6), 126.5 (d, 3JC–P 11.3, C4), 125.9 (s, C5), 118.8 (s, C7), 47.8 (s, N(CH3)2 on 2C8), 43.9 (s, N(CH3)2 on 2C1). 31P{1H} NMR (146 MHz, CDCl3) δP (5c): 64.22. IR (hexane) cm−1 : 2046 (sharp), 1971 (m, br), 1944 (s, br), 1935 (s, br). ESI-MS (MeCN, formic acid) m/z: 399.1 [3 + H]+, 567.1 [5c + H]+.
ESI-MS (5a; CH2Cl2–HBF4) 483 (100%) [PC32H68]+, (5b; CH2Cl2–HBF4) 521 (23%) [5b + H]+, 483 (100%) [PC32H68]+, 369 (51%) [PO(C6H4OMe)3H]+, 353 (23%) [P(C6H4OMe)3 + H]+, (5c; CH2Cl2–HCl) 567 (100%) [5c + H]+, 483 (20%) [PC32H68]+, 415 10% [3O + H]+, 399 5% [3 + H]+.
ESI-MS (CH2Cl2–formic acid) 723 [7 + H]+, 399 [3 + H]+. IR ν(CO) (CH2Cl2–hexane) (cm−1) 2070 (s), 1978, 1920 (br), 1860 (w).
Crystal structure determinations
X-Ray crystallographic data for 3 and 3·HBr·C2H5OH were collected in the X-ray Laboratory of the Cambridge University Chemical Laboratory. Measurements were made on a Nonius Kappa CCD diffractometer at −93 °C with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Data for 4 and 5c were collected at −80 °C on a Bruker PLATFORM/SMART 1000 CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å), at the X-ray Crystallography Laboratory of the University of Alberta. The structures were solved by direct methods, except for 5c (Patterson search/structure expansion)22 and refined by full-matrix least-squares on F2.23 A Flack parameter24 of 0.14 (11) for 4 indicated a small degree of racemic twinning, which was accommodated during the refinement. ORTEP diagrams were generated using ORTEP 325 and POV-Ray 3.5.26Crystal data and selected details of structure determinations are given in Table 1.
| 3 | 3·HBr·C2H5OH | 4 | 5c | |
|---|---|---|---|---|
| Formula | C26H27N2P | C28H33BrN2OP | C34H39N4P | C30H27FeN2O4P |
| M r | 398.47 | 524.44 | 534.66 | 566.36 |
| Crystal dimensions/mm | 0.46 × 0.21 × 0.12 | 0.46 × 0.10 × 0.05 | 0.64 × 0.24 × 0.15 | 0.62 × 0.41 × 0.37 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/n | P21/c | Pna21 | P21/n |
| a/Å | 17.1220(3) | 16.0283(3) | 15.4223(12) | 10.5700(9) |
| b/Å | 10.1292(2) | 7.18140(10) | 10.8193(8) | 15.0412(13) |
| c/Å | 25.3141(5) | 22.5380(6) | 17.8969(14) | 17.6505(16) |
| β/° | 93.8090(10) | 98.9630(10) | 90 | 104.2580(11) |
| V/Å3 | 4380.58(14) | 2562.57(9) | 2986.3(4) | 2719.7(4) |
| Z | 8 | 4 | 4 | 4 |
| D c/g cm−3 | 1.208 | 1.359 | 1.189 | 1.383 |
| µ/mm−1 | 0.140 | 1.690 | 0.121 | 0.652 |
| T/°C | −93(2) | −93(2) | −80 | −80 |
| θ Range/° | 3.58 to 27.51 | 3.66 to 27.51 | 2.20 to 25.85 | 2.46 to 26.37 |
| Total data | 32696 | 23406 | 22819 | 16008 |
| hkl Ranges | −22 to 22, −13 to 13, −32 to 32 | −20 to 20, −9 to 9, −29 to 29 | −19 to 19, −13 to 13, −22 to 22 | −13 to 13, −18 to 18, −22 to 21 |
| Independent reflections | 10020 | 5838 | 6154 | 5553 |
| R int | 0.0831 | 0.0613 | 0.0504 | 0.0224 |
| Range transmission factors | 0.984–0.848 | 0.990–0.884 | 0.9821–0.9265 | 0.7945–0.6881 |
| Data/restraints/parameters | 10020/0/523 | 5838/0/299 | 6154/0/353 | 5553/0/343 |
| Goodness-of-fit (S) | 1.023 | 1.038 | 1.023 | 1.021 |
| R 1 [Fo2 ≥ 2σ(Fo2)] | 0.0547 | 0.0546 | 0.0503 | 0.0368 |
| wR 2 [Fo2 ≥ 3σ(Fo2)] | 0.1137 | 0.1170 | 0.1374 | 0.1085 |
| Largest diff. peak, hole/e Å−3 | 0.265, −0.343 | 0.684, −0.542 | 0.234, −0.248 | 0.549, −0.595 |
CCDC reference numbers 611411–611414.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b609561e
Results and discussion
Several groups including those of Alder,27 Staab,28 Pozharskii29 and Lloyd-Jones30 have investigated and derivatised 1. Numerous kinetic,31 structural,32 spectroscopic33 and theoretical investigations34 of 1 and its derivatives have also been carried out. We investigated a variety of bromination methods as a route to functionalisation of 1; nearly all gave mixtures of mono and dibrominated products; HBr (48%, aq.) in AcOH–DMSO,35 Br2 in CCl436 or H2SO4,37 TBABr3 in CH2Cl238 and 2,4,4,6-tetrabromo-2,5-cyclohexadien-1-one in CH2Cl2.39 In our hands, the best brominating system proved to be N-bromosuccinimide (NBS) in THF at −78 °C, being regiospecific for monobromination at the 2-position, producing 1,8-bis(dimethylamino)-2-bromonaphthalene (2) in good yield.40 Little polybromination was observed under these conditions, indicating either that the system is under kinetic control or that the NMe2 group ortho- to the position of bromination participates in the bromination through an unknown mechanism.Syntheses of 3 and 4 from 2 were straightforward via lithiation followed by treatment with PPh(3−n)Cln (n = 1 or 2, see Scheme 1) but purification proved to be more complicated. Proton sponge derivatives adhered strongly to both silica and sand and smeared on neutral activated alumina. Low loadings of these compounds could be partially purified by alumina column chromatography, a general method involved loading the compound in CH2Cl2 and using 19 : 1 hexane–THF with trace triethylamine (TEA) to elute. Distillation proved to be the method of choice to purify multigram quantities of 2 but could not be easily used to separate different (ortho/para) isomers of 2. In general, separation of different proton sponge derivatives from each other was difficult—for example, 1 and 3 are both soluble in 2—and therefore clean reactions were important. The nature of the compounds meant that incidental protonation during workup procedures had to be considered since this altered the physical properties of the compounds. Intentional protonation/deprotonation could however be used to aid purification. Standard organic workup techniques (extractions, washing, drying with MgSO4) and chromatography were used initially but eventually avoided where possible as they greatly reduced yields. The bulky phosphines 3 and 4 proved to be fairly air stable when solid, and were stable in deoxygenated solution under N2. They could be reversibly protonated/deprotonated with strong acid and base multiple times without decomposition. The phosphine oxides (which were also yellow) could be removed by stirring the impure phosphine in minimal EtOH under N2, cooling on ice and filtering, thus removing the soluble oxide.
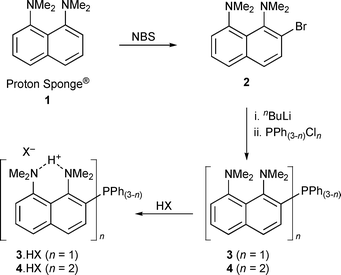 | ||
| Scheme 1 Syntheses of 2–4 from Proton Sponge® (1). | ||
X-Ray crystal structures of both 3 and its hydrobromide, 3·HBr (along with an ethanol of crystallisation) were obtained (Fig. 1 and 2). Protonation of 3 to give 3·HBr significantly increases the planarity of the naphthalene; the average deviation from the best-fit plane defined by the ten naphthalene carbon atoms is 0.102 Å for 3 but only 0.007 Å for 3·HBr. The deviation from planarity is also manifested by the N–C(1)–C(8)–N torsion angles, 37.4° for 3 but just 1.2° for 3·HBr. The twisting of the naphthalene is consistent with co-repulsion of the lone pairs, relief of this strain being the suggested driving force for protonation.12 The position of the nitrogen lone pairs can be estimated from the orientation of the NMe2 methyl groups. Upon protonation the N⋯N distance is reduced from 2.90 to 2.56 Å (cf. 2.55–2.62 Å in protonated 141) and as the nitrogen lone pairs coordinate to the proton there is less involvement with the naphthalene π system resulting in longer C(1)–N(1) and C(8)–N(2) bonds. The average length of these bonds is 1.46 Å in 3·HBr and 1.43 Å in 3. The sum of angles around N(1) and N(2) in 3 are 355° (∼98% sp2 character) and 338° (∼94% sp2 character), respectively. In 3·HBr the nitrogen atoms become marginally more sp3 like, as the angles reduce to 343° (95%) and 337° (93%). The anisotropic displacement parameters for N(1) and N(2) are smaller in 3·HBr than in 3, reflecting the increased bonding and reduced freedom of the nitrogen atoms.
 | ||
| Fig. 1 ORTEP plot of 3 (one of the two independent molecules in the unit cell; bond lengths and angles do not vary significantly between the two). Non-hydrogen atoms are represented by Gaussian ellipsoids at the 50% probability level. Hydrogen atoms are shown with arbitrarily small thermal parameters. Key bond lengths (Å) and angles (°): P1–C2 1.842(2), N1–C1 1.428(2), N2–C8 1.430(3); C2–P1–C21 100.54(9), C2–P1–C31 100.62(9), C21–P1–C31 104.15(10). | ||
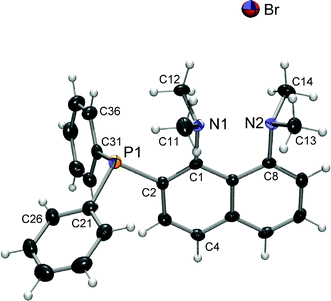 | ||
| Fig. 2 ORTEP plot of 3·HBr (ethanol of crystallisation not shown). Non-hydrogen atoms are represented by Gaussian ellipsoids at the 50% probability level. Hydrogen atoms are shown with arbitrarily small thermal parameters. Key bond lengths (Å) and angles (°): P1–C2 1.857(3), N1–C1 1.459(3), N2–C8 1.468(4); C2–P1–C31 100.79(13), C2–P1–C31 101.70(14), C21–P1–C31 104.64(14). | ||
The diproton sponge phosphine ligand 4 was also examined structurally (Fig. 3). One of the proton sponge groups on 4 has similar geometry to its counterpart in 3; an N–C1–C8–N torsion angle of 36.6°, an average deviation from the best-fit plane defined by the ten naphthalene carbon atoms of 0.098 Å, and an N–N distance of 2.91 Å. However, the other proton sponge group attached to the phosphorus (containing N1 and N2) is much less distorted from planarity, with the respective torsion angle, deviation from planarity and N–N distance being 16.55°, 0.057 Å and 2.84 Å. The reason for the difference is not immediately obvious, but presumably can be attributed to crystal packing forces and suggests a reasonable degree of flexibility of the proton sponge unit.
 | ||
| Fig. 3 ORTEP plot of 4. Non-hydrogen atoms are represented by Gaussian ellipsoids at the 50% probability level. Hydrogen atoms are shown with arbitrarily small thermal parameters. Key bond lengths (Å) and angles (°): P–C12 1.843(3), P–C22 1.850(3), N1–C11 1.413(5), N2–C19 1.432(4), N3–C21 1.423(4), N4–C29 1.410(4); C12–P–C32 100.45(12), C12–P–C31 100.67(12), C22–P–C31 101.30(12). | ||
Ligands 3 and 4 both display pH-dependent solubility. In their unprotonated form they dissolve readily in organic solvents such as CH2Cl2 or THF, but addition of acid greatly increases solubility in polar solvents; 3·HBF4 is water-soluble, for example. Neutralisation with a strong base such as NaOH results in the ligand moving back into the organic phase. Ligands that confer water-solubility to their complexes, such as the well-known 1,3,5-triaza-7-phosphaadamantane (PTA),42 have numerous applications in aqueous phase or biphasic homogeneous catalysis.
Transprotonation experiments
The high basicity of proton sponges has made determining their pKa values challenging, the pKa of the solvent in which the study is conducted being a constraining factor. Temperature-jump43 or NMR methods44 have been used by other groups. We adopted the latter approach, albeit less accurate, due to the very clear signal of the coordinated proton in the 1H NMR spectra at high chemical shifts (∼20 ppm) for all derivatives of 1. Solubility of all four compounds (the two protonated/unprotonated pairs) in the deuterated solvent was a critical issue since any insolubility would affect the equilibrium and therefore the determined value. HBF4 and CD3CN proved to be the best combination. Equimolar amounts of 1 and 3·HBF4 were dissolved in CD3CN and the 1H NMR spectrum was recorded. The same experiment was conducted with equimolar amounts of 1·HBF4 and 3. In both cases, equilibrium between 3·HBF4 and 1·HBF4 was quickly reached and the ratio of these species remained roughly constant from within 5 min of sample mixing. 1H NMR signals in the low-field region corresponded to the proton coordinated by the different proton sponges, δ 19.7 ppm corresponding to 3·HBF4 and δ 18.7 ppm to 1·HBF4. The ratios of the protonated species were averaged over 4 samples and the first pKaH of 3 (CD3CN) was calculated as 18.24 ± 0.02 (1 std) units at 297 K, demonstrating a slight increase in basicity when compared to the value for 1 (18.18 in CH3CN).20Fig. 4 shows the relative ratios of the two peaks. | ||
| Fig. 4 1H NMR spectra of an equimolar mixture of 1 and 3·HBF4 (an identical spectrum is obtained for an equimolar mixture of 1·HBF4 and 3) in the low-field region. | ||
The slightly higher pKaH is in agreement with the observation that proton sponges with “buttressing” substituents in the 2- and 7-positions demonstrate increased pKaH values as the lone pairs are forced closer together in the unprotonated state, the relief of strain upon protonation being correspondingly higher.12,45
After determining that the equilibrium set up was identical from either starting point, the experiment was repeated using equimolar quantities of 4 and 1·HBF4. The low solubility of 4 in CD3CN necessitated very dilute solutions to ensure total dissolution and no precipitation of any species during the experiment, 1H NMR experiments were run overnight (15000 scans). 4·HBF4 showed a single peak in the 1H NMR spectra at 19.9 ppm (relative integral 1). The proton coordinated by 1 gave a peak at 18.7 ppm (relative integral 0.6). Due to the structural similarity between 3 and 4 it is anticipated that their first pKaH will be approximately equal. Since the combined coordination by both proton sponge sites of 4 is demonstrated by the integrals to be less than two-thirds, this implies that the second pKaH of 4 is significantly less than the first pKaH of 1. This is not unexpected, as the second pKaH of 4 corresponds to the protonation of a formally charged compound, 4·HBF4, to form 4·2HBF4.
ESI-MS of the proton sponge derivatives
Throughout this work we have observed that ESI-MS of proton sponge derivatives generally show no sign of cationisation by species other than H+, even in the presence of excess Li+, Na+, K+ or NH4+. The exception is the spectrum of 5c, which like 5a and 5b, provides an [M + Na]+ signal, though this is certainly due to interactions with the carbonyl ligands rather than the with the proton sponge.6b ESI-MS of a solution of a 1 : 1 molar ratio of tetradecyltrihexylphosphonium bis(trifluromethylsulfonyl)amide and the ligand 3 showed that the ionisation efficiency of 3 approaches that of the formally charged phosphonium ion (in CH2Cl2, CH3CN or MeOH in presence of a H+ source). This has important implications with the use of the ligand to identify catalytic intermediates; reactions typically employ catalysts at a 1–10 mol% concentration1a and so the concentration of catalytic intermediates can be expected to be very much lower. Therefore the ionisation efficiency of the ligand must be as high as possible for the ligand to reveal these low concentration reactive intermediates in the ESI mass spectrum. Even without added H+, the ligand 3 provides a very strong signal from a dichloromethane solution, approximately 50% of the intensity of the phosphonium ion (Fig. 5).![Positive-ion ESI-MS of an equimolar mixture of 3 and an internal reference, [P(C6H13)3(C14H29)]+, in CH2Cl2. The dotted line indicates the change in intensity of the [3 + H]+ signal observed when excess H+ (a drop of formic acid) is deliberately added to the solution.](/image/article/2006/DT/b609561e/b609561e-f5.gif) | ||
| Fig. 5 Positive-ion ESI-MS of an equimolar mixture of 3 and an internal reference, [P(C6H13)3(C14H29)]+, in CH2Cl2. The dotted line indicates the change in intensity of the [3 + H]+ signal observed when excess H+ (a drop of formic acid) is deliberately added to the solution. | ||
Metal complexes
Several organometallic complexes incorporating 3 as a ligand were prepared by standard methods (Scheme 2).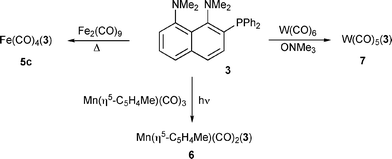 | ||
| Scheme 2 Syntheses of 5c, 6 and 7 from 3. | ||
The preparations of 5c, 6 and 7 were adapted from syntheses of Fe(CO)4(PPh3),46 Mn(η5-C5H4Me)(CO)2(PPh3),47 and W(CO)5(PPh3),48 respectively. For the sake of comparison, the known complexes Fe(CO)4(PR3) (R = Ph, 5a; R = p-C6H4OMe, 5b) were also prepared.
5c readily crystallised and an X-ray crystal structure was obtained of this complex (Fig. 6). It revealed a slightly distorted trigonal-bipyramidal geometry about iron with the phosphine in an axial position, as is seen in the analogous compound Fe(CO)4(PPh3) (5a).49 Although it is often assumed that in trigonal bipyramidal organometallic complexes the better π-acceptor ligand adopts an equatorial position,50 evidence points to the σ-donation also being important51 with the weaker σ-donor ligand favouring the equatorial position (where CO < PPh3 in terms of σ-donation).52 Steric effects would also be expected to encourage the largest ligand in the complex to occupy the equatorial position (with only two groups at 90° cf. three for substituents in an axial position). The orientation of the N(1) methyl groups suggests that there may be a weak interaction between the N(1) lone pair and the Fe centre, but in fact the intramolecular distance of 3.58 Å is substantially longer than the sum of van der Waals radii.
 | ||
| Fig. 6 ORTEP plot of 5c. Non-hydrogen atoms are represented by Gaussian ellipsoids at the 50% probability level. Hydrogen atoms are shown with arbitrarily small thermal parameters. Key bond lengths (Å) and angles (°): Fe–P 2.2739(6), Fe–C1 1.791(2), Fe–C4 1.776(3), P–C12 1.8416(19), N1–C11 1.428(2), N2–C19 1.429(3); Fe–P–C12 117.46(6), C31–P–C41 101.38(9), C12–P–C41 103.95(9). | ||
Complexes 5a, 5b and 5c exhibited carbonyl stretches in the IR region in the order of PPh3 > P(C6H4OMe)3 ∼ 3 (2051, 2047, 2046 cm−1 as highest-frequency A′ band) indicating that the net electron density donated to the iron by 3 is greater than for the other ligands, with back-donation to the CO ligands in this complex being correspondingly greater, weakening the CO bond.
Just as is the case for the other unprotonated sponges, the 1,8-bis(dimethylamino)naphthyl group is significantly distorted from planarity, with an N–C1–C8–N torsion angle of 28.1°, an average deviation from the best-fit plane defined by the ten naphthalene carbon atoms of 0.078 Å, and an N–N distance of 2.948 Å. These parameters are middle-of-the-range compared to 3 and 4, with the exception of the N–N distance, which is significantly longer than the other examples. Again, this appears to point to a reasonable degree of flexibility in the 1,8-bis(dimethylamino)naphthyl group and to the participation of crystal packing forces in the structure. The phosphine ligand in 5c has a cone angle53 of 168° taking into account the surfaces of the van der Waals spheres in the substituents,54 with measurements taken directly from the crystal structure. The cone angle of PPh3 in the triphenylphosphine analogue Fe(CO)4PPh3 (5a) is 151° when calculated by the same method.55
Comparison of electrospray activity of 5a, 5b and 5c
Using [P(C6H13)3(C14H29)]+ as an internal reference or in competition experiments it was shown that the ionisation efficiency of 5c is approximately 20x that of 5b in acidic CH2Cl2, despite 5c having just one site for protonation compared to three for 5c. 5a was not detectable under these conditions. The 20x improvement in signal intensity for 5c over 5b is probably conservative; the mass spectra of 5c were greatly superior under nearly all conditions but less pronounced under the relatively high concentrations used in the competition and internal reference experiments. It certainly appears that the primary advantage of the proton sponge phosphine will be under conditions that are marginal, and those in which the speciation is complicated. Other circumstances in which ESI-MS analysis is challenging and where proton sponge phosphines may provide useful handles include the characterisation of complexes in ionic liquids56 or in non-polar solvents such as toluene or hexane (in which lipophilic ionic liquids are required to assist the formation of a stable spray).57The proton sponge ligand is also more selective, always providing only [M + H]+ ions except in neutral or basic solutions containing added Na+, which presumably associates with the complex via an interaction with the carbonyl ligands. Both 5b and 5c showed the appearance of the free ligand in all spectra; the presence of phosphine oxide suggests that the ligand is not appearing as a fragment generated by the ionisation process, but rather is dissociated to a reasonable degree in solution (Fig. 7). Its appearance suggests a degree of decomposition; this is not unusual in ESI mass spectra, as the concentration of sample used is very low (micromolar) and so the presence of small amounts of air and moisture in the solvent becomes significant. The 31P NMR spectrum, collected at much higher concentration, showed insignificant levels of decomposition (in the form of free phosphine or phosphine oxide).
 | ||
| Fig. 7 Positive-ion ESI-MS of 5c in CH2Cl2–trace formic acid. The inset shows the experimental and calculated isotope patterns. Free ligand 3 and free oxidised ligand 3a were always present in spectra of metal complexes to at least some degree. | ||
The syntheses of the manganese and tungsten complexes 6 and 7 further demonstrated the ability of this proton sponge derivative 3 to act as a conventional phosphine ligand, with bonding via phosphorus and the –NMe2 groups of the proton sponge uninvolved in bonding to the metal. Both complexes displayed strong [M + H]+ ions in their ESI mass spectra. MS/MS studies on these compounds (and of 5c) revealed that the primary fragmentation process was loss of the protonated ligand from the metal core (see Fig. 8). For 6, a small amount of [6 − 2CO + H]+ is present, indicating competition between CO and phosphine loss; this process is not observed at all for 5c or 7.
![Positive-ion ESI-MS/MS of [6 + H]+ (top) and [7 + H]+ (bottom) in CH2Cl2/trace formic acid. Collision voltage was set to reduce the parent ion intensity to ∼50%. In both cases, fragmentation involves loss of the protonated ligand from the metal; the ligand retains the charge.](/image/article/2006/DT/b609561e/b609561e-f8.gif) | ||
| Fig. 8 Positive-ion ESI-MS/MS of [6 + H]+ (top) and [7 + H]+ (bottom) in CH2Cl2/trace formic acid. Collision voltage was set to reduce the parent ion intensity to ∼50%. In both cases, fragmentation involves loss of the protonated ligand from the metal; the ligand retains the charge. | ||
Conclusions
In order to detect neutral complexes by ESI-MS under challenging conditions of low concentration or in the presence of abundant competing species-circumstances common when wishing to study catalytic systems, for example—the complex must possess functionality capable of selective ionisation with high efficiency. Proton sponges confer exactly these properties to a ligand, and the ligand in turn confers them to the complexes to which they are bound. Absolute selectivity for H+ means simple spectra uncomplicated by multiple signals derived from different ionisation mechanisms. Complexes incorporating proton sponge phosphines provide strong [M + H]+ ions even in solutions free of added H+ or protic solvents, making the ligands ideal for facilitating direct ESI-MS analysis of catalytic reactions involving neutral complexes and phosphine ligands.Acknowledgements
N. J. F. thanks the EPSRC for a studentship and Professor Brian F. G. Johnson for support. J. S. M. thanks Natural Sciences and Engineering Research Council (NSERC) of Canada, the Canada Foundation for Innovation (CFI) and the British Columbia Knowledge Development Fund (BCKDF), and the University of Victoria for instrumentation and operational funding.References
- (a) P. Chen, Angew. Chem., Int. Ed., 2003, 42, 2832 CrossRef CAS; (b) R. A. J. O'Hair, Chem. Commun., 2006, 1469 RSC.
- W. Henderson and J. S. McIndoe, Mass Spectrometry of Inorganic and Organometallic Compounds: Tools, Techniques and Tips, John Wiley & Sons, Chichester, 2005 Search PubMed.
- C. Evans and W. Henderson, Inorg. Chim. Acta, 1999, 294, 183 CrossRef CAS.
- Y. Gimbert, D. Lesage, A. Milet, F. Fournier, A. E. Greene and J. C. Tabet, Org. Lett., 2003, 5, 4073 CrossRef CAS.
- G. J. van Berkel and F. Zhou, Anal. Chem., 1995, 67, 3958 CrossRef CAS.
- (a) W. Henderson, J. S. McIndoe, B. K. Nicholson and P. J. Dyson, Chem. Commun., 1996, 1183 RSC; (b) W. Henderson, J. S. McIndoe, B. K. Nicholson and P. J. Dyson, J. Chem. Soc., Dalton Trans., 1998, 519 RSC.
- W. Henderson and B. K. Nicholson, J. Chem. Soc., Chem. Commun., 1995, 2531 RSC.
- C. Decker, W. Henderson and B. K. Nicholson, J. Chem. Soc., Dalton Trans., 1999, 3507 RSC.
- C. Evans and B. K. Nicholson, J. Organomet. Chem., 2003, 665, 95 CrossRef CAS.
- (a) H. A. Staab and T. Saupe, Angew. Chem., Int. Ed. Engl., 1988, 27, 865 CrossRef; (b) A. L. Llamas-Saiz, C. Foces-Foces and J. Elguero, J. Mol. Struct., 1994, 328, 297 CrossRef.
- R. W. Alder, P. S. Bowman, W. R. S. Steele and D. R. Winterman, Chem. Commun., 1968, 723 RSC.
- R. W. Alder, Chem. Rev., 1989, 89, 1215 CrossRef CAS.
- A. F. Pozharskii, Russ. Chem. Rev., 1998, 67, 1 RSC.
- S. N. Gamage, R. H. Morris, S. J. Rettig, D. C. Thackeray, I. S. Thorburn and B. R. James, J. Chem. Soc., Chem. Commun., 1987, 894 RSC.
- R. P. Hughes, I. Kovacik, D. C. Lindner, J. M. Smith, S. Willemsen, D. Zhang, I. A. Guzei and Arnold L. Rheingold, Organometallics, 2001, 20, 3190 CrossRef CAS.
- A. Di Saverio, F. Focante, I. Camurati, L. Resconi, T. Beringhelli, G. D'Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Inorg. Chem., 2005, 44, 5030 CrossRef CAS.
- T. Yamasaki, N. Ozaki, Y. Saika, K. Ohta, K. Goboh, F. Nakamura, M. Hashimoto and S. Okeya, Chem. Lett., 2004, 33, 928 CrossRef CAS.
- F. Terrier, J.-C. Halle, M.-J. Pouet and M.-P. Simonnin, J. Org. Chem., 1986, 51, 409 CrossRef CAS.
- T. Saupe, C. Krieger and H. A. Staab, Angew. Chem., Int. Ed. Engl., 1986, 25, 451 CrossRef.
- V. Raab, J. Kipke, R. M. Gschwind and J. Sundermeyer, Chem.–Eur. J., 2002, 8, 1682 CrossRef.
- J. A. S. Howell, M. G. Palin, P. McArdle, D. Cunningham, Z. Goldschmidt, H. E. Gottlieb and D. Hezroni-Langerman, Inorg. Chem., 1993, 32, 3493 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467 CrossRef.
- G. M. Sheldrick, SHELXL-93. Program for crystal structure determination, University of Göttingen, Germany, 1993 Search PubMed.
- H. D. Flack, Acta Crystallogr., Sect. A, 1983, 39, 876 CrossRef; H. D. Flack and G. Bernardinelli, Acta Crystallogr., Sect. A, 1999, 55, 908 CrossRef; H. D. Flack and G. Bernardinelli, J. Appl. Crystallogr., 2000, 33, 1143 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- Available online at http://www.povray.org/.
- R. W. Alder, M. R. Bryce, N. C. Goode, N. Miller and J. Owen, J. Chem. Soc., Perkin Trans. 1, 1981, 2840 RSC.
- H. A. Staab, C. Krieger, G. Hieber and K. Oberdorf, Angew. Chem., Int. Ed. Engl., 1997, 36, 1884 CAS.
- A. F. Pozharskii, O. V. Ryabtsova, V. A. Ozeryanskii, A. V. Degtyarev, O. N. Kazheva, G. G. Alexandrov and O. A. Dyachenko, J. Org. Chem., 2003, 68, 10109 CrossRef CAS.
- J. P. H. Charmant, G. C. Lloyd-Jones, T. M. Peakman and R. L. Woodward, Eur. J. Org. Chem., 1999, 2501 CrossRef CAS.
- F. Hibbert, Acc. Chem. Res., 1984, 17, 115 CrossRef CAS.
- (a) F. Hibbert and J. Emsley, Adv. Phys. Org. Chem., 1990, 26, 255 CAS; (b) M. R. Truter and B. L. Vickery, J. Chem. Soc., Dalton Trans., 1972, 395 RSC; (c) H. Einspahr, J. B. Robert, R. E. Marsh and J. D. Roberts, Acta. Crystallogr., Sect. B, 1973, 29, 1611 CrossRef CAS.
- M. Pietrzak, J. Wehling, H. Limbach and R. M. Claramunt, J. Am. Chem. Soc., 2001, 123, 4338 CrossRef CAS.
- J. A. Platts, S. T. Howard and K. Wozniak, J. Org. Chem., 1994, 59, 4647 CrossRef CAS.
- G. Majetich, R. Hicks and S. Reister, J. Org. Chem., 1997, 62, 4321 CrossRef CAS.
- R. Adams and C. S. Marvel, Org. Synth., 1941, I, 128.
- N. V. Vistorobskii and A. F. Pozharskii, Zh. Org. Khim., 1989, 25, 2154 CAS.
- J. Berthelot, C. Guette, M. Essayegh, P. L. Desbene and J. J. Basselier, Synth. Commun., 1986, 16, 1641 CrossRef CAS.
- G. J. Fox, G. Hallas, J. D. Hepworth and K. N. Paskins, Org. Synth., 1976, 55, 181.
- H. M. Gilow and D. E. Burton, J. Org. Chem., 1981, 46, 2221 CrossRef CAS.
- (a) D. E. Fenton, M. R. Truter and B. L. Vickery, J. Chem. Soc. D, 1971, 93 RSC; (b) D. Pyzalska, R. Pyzalski and T. Borowiak, J. Crystallogr. Spectrosc. Res., 1983, 13, 211 CAS.
- A. D. Phillips, L. Gonsalvi, A. Romerosa, F. Vizza and M. Peruzzini, Coord. Chem. Rev., 2004, 248, 955 CrossRef CAS.
- F. Hibbert, J. Chem. Soc., Perkin Trans. 2, 1974, 1862 RSC.
- (a) R. W. Alder, P. Eastment, N. M. Hext, R. E. Moss, A. G. Orpen and J. M. White, J. Chem. Soc., Chem. Commun., 1988, 1528 RSC; (b) M. A. Zirnstein and H. A. Staab, Angew. Chem., Int. Ed. Engl., 1987, 26, 460 CrossRef.
- F. Hibbert and K. P. P. Hunte, J. Chem. Soc., Perkin Trans. 2, 1983, 1895 RSC.
- A. F. Clifford and A. K. Mukherjee, Inorg. Synth., 1966, 8, 185 CAS.
- A. J. Arduengo, M. Lattman, H. V. Rasika Dias, J. C. Calabrese and M. Kline, J. Am. Chem. Soc., 1991, 113, 1799 CrossRef.
- L. Hirsivaara, L. Guerricabeitia, M. Haukka, P. Suomalainen, R. H. Laitinen, T. A. Pakkanen and J. Pursiainen, Inorg. Chim. Acta, 2000, 307, 47 CAS.
- P. E. Riley and R. E. Davis, Inorg. Chem., 1980, 19, 159 CrossRef CAS.
- S. A. Goldfield and K. N. Raymond, Inorg. Chem., 1974, 13, 770 CrossRef CAS.
- A. R. Rossi and R. Hoffmann, Inorg. Chem., 1975, 14, 365 CrossRef CAS.
- L. R. Martin, F. W. B. Einstein and R. K. Pomeroy, Inorg. Chem., 1983, 22, 1961 CrossRef.
- C. A. Tolman, Chem. Rev., 1977, 77, 31.
- D. M. P. Mingos and T. E. Muller, J. Organomet. Chem., 1995, 500, 251 CrossRef CAS.
- T. E. Muller and D. M. P. Mingos, Transition Met. Chem., 1995, 20, 533 CrossRef.
- (a) P. J. Dyson, J. S. McIndoe and D. Zhao, Chem. Commun., 2003, 508 RSC; (b) P. J. Dyson, I. Khalaila, S. Luettgen, J. S. McIndoe and D. Zhao, Chem. Commun., 2003, 2204 RSC.
- M. A. Henderson and J. S. McIndoe, Chem. Commun., 2006, 2872 RSC.
Footnotes |
| † This paper is dedicated to the memory of Dr Alex Hopkins (1975–2006). |
| ‡ The HTML version of this article has been enhanced with colour images. |
| This journal is © The Royal Society of Chemistry 2006 |