Cation and anion diversity in [M(dithiooxalate)2]2− salts: structure robustness in crystal synthesis†
Christopher J.
Adams
,
Paul C.
Crawford
,
A.
Guy Orpen
* and
Thomas J.
Podesta
School of Chemistry, University of Bristol, Bristol, UK BS8 1TS. E-mail: Guy.Orpen@bris.ac.uk
First published on 12th July 2006
Abstract
A series of crystalline salts based on the [M(dto)2]2− (dto = 1,2-dithiooxalate, M = Ni, Pt, Cu) dianion with hydrogen-bond donor cations have been synthesised following a molecular tectonics approach. The chelating M(dto)⋯HN supramolecular synthon has been exploited in a systematic study of its robustness. The effects of competition between hydrogen-bond acceptors, of the shape and functionality of the cations and of varying the metal in the anion are discussed. The preparation and structural characterisation of the new crystalline phases [4,4′-H2bipy][Pt(dto)2] (2), [HNC5H4CO2H-4]2[Pt(dto)2] (5), [HNC5H4CO2H-3]2[Pt(dto)2] (6), [HNC5H4CH2CO2H-4]2[Ni(dto)2] (7), [HNC5H4CH2CO2H-3]2[Ni(dto)2] (8), [HNC5H4CONH2-4]2[Ni(dto)2] (9), [HNC5H4CHNOH-4]2[Ni(dto)2] (10), [HNC5H4CHNOH-3]2[Ni(dto)2] (11), [4,4′-H2bipip][Ni(dto)2] (12), [H2NC5H9CO2H-4]2[Pt(dto)2] (14), [H2NC5H9CO2H-4]2[Cu(dto)2] (15), [H2NC5H9CO2H-3]2[Ni(dto)2][H2O]2 (16), [H2NC5H9CO2H-3]2[Pt(dto)2][H2O]2 (17), [H2NC5H9CO2H-3]2[Cu(dto)2][H2O]2 (18), [H(Me)NC5H9CO2H-4]2[Ni(dto)2][H2O]2 (20) is reported. The charge-assisted NH⋯dto synthon is formed in each of compounds 1–20, and is apparently much more robust than the conventional synthons used (such as the carboxylic acid dimer), which have a much lower rate of occurrence. The NH⋯dto synthon may be generalised to 3- and 4-pyridinium species and 3- and 4-piperidinium derivatives. In the latter cases branching of the hydrogen-bond networks through the NH2 groups arises. The robustness of the NH⋯dto synthon allows structures of the form [NH cation]2[M(dto)2] to be regarded as being formed by the packing of neutral supermolecules. Cases of isomorphism (as in 16–18) and latent polymorphism (e.g. in 4 and 6) are noted.
Introduction
The designed synthesis of molecular crystal structures—a field which might be termed synthetic crystallography—is a key goal in modern chemistry. One approach to its achievement is the use of molecular recognition interactions (termed supramolecular synthons by Desiraju)1 to bind the individual molecular components (termed tectons by Wuest and others)2 of a structure into a desired periodic pattern. Once identified, the (supramolecular) synthon may be exploited through the synthesis of appropriate molecular tectons which incorporate complementary recognition or binding sites. A corollary of this approach is that the synthons must be robust, i.e. they can be relied upon to form when the required functional groups are present in the tectons used, even in the face of competition. This is the analogue in synthetic crystallography of functional group tolerance in the field of molecular synthesis. Furthermore it is desirable that the tectons exhibit sufficient diversity that a substantial range of related or analogous crystal structures may be prepared. If this is possible then “genuine” crystal engineering may be achievable—that is to say, the planned construction, modification or adjustment of crystal structures to afford desired properties and functions—e.g. of a particular metric or chemical, electronic or magnetic type.In previous work we and others have followed this approach, exploring the charge assisted hydrogen-bonding interactions of anionic perhalometallate hydrogen-bond acceptor tectons such as A, with cationic, often pyridinium, NH donors, to prepare inorganic–organic salt crystal structures incorporating the NH⋯Cl2M interaction B and related synthons.3,4 We have also reported the utilisation of more complex anionic tectons including tetracyanometallate, [M(mnt)2]2− (mnt = 1,2-dithiomaleonitrile) and tetrathiocyanometallate dianions.5
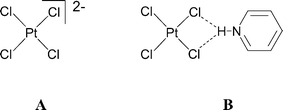
Similar approaches, also exploiting charge-assisted hydrogen-bonding, have been used by others, including Brammer, Hosseini, Schröder, Das and their co-workers,4,6,7 to exploit metal complex anions as hydrogen-bond acceptors, while Aakeröy, Braga, Ward and their co-workers, among others, have developed related systems with organic anions.8,9
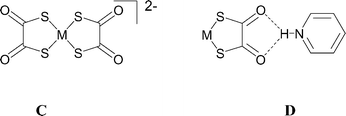
We have briefly reported the use of the [Ni(dto)2]2− (dto = 1,2-dithiooxalate) dianionic tecton C (with M = Ni) as a hydrogen-bond acceptor.10 The chelating pyridinium⋯dto synthon D was shown to be an effective mimic for B in salts of [Ni(dto)2]2− with pyridinium and piperidinium NH hydrogen-bond donors, forming synthon D in each of a series of five salts.10 For example, the supramolecular structure of [4,4′-bipyridinium][Ni(dto)2], 1, contains a hydrogen-bonded ribbon motif, analogous to that found in [4,4′-bipyridinium][PtCl4], as shown in Fig. 1.
![The hydrogen-bond ribbon motifs found in the crystal structures of [4,4′-H2bipy][Ni(dto)2], 1 (below)10 and [4,4′-H2bipy][PtCl4]3c (above).](/image/article/2006/DT/b604319d/b604319d-f1.gif) | ||
| Fig. 1 The hydrogen-bond ribbon motifs found in the crystal structures of [4,4′-H2bipy][Ni(dto)2], 1 (below)10 and [4,4′-H2bipy][PtCl4]3c (above). | ||
Dianions of form C offers a suitable combination of good hydrogen-bond acceptor ability (through the carbonyl groups), high negative charge (1− per dto site), inertness towards ligand substitution (as a result of metal chelation through the sulfur atoms), and the absence of any hydrogen-bond donor functionalities. Aside from crystal engineering studies, [M(dto)2]2− anions have been used as ligands in coordination chemistry.11
In order to probe the robustness of the M(dto)⋯HN synthon D and its utility in diversifying the crystal structures it may be used to prepare, we have sought to synthesise salts of a range of [M(dto)2]2− dianions (M = Ni, Pt, Cu) with various molecular and supramolecular cations based on pyridinium and piperidinium derivatives.
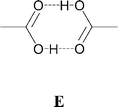
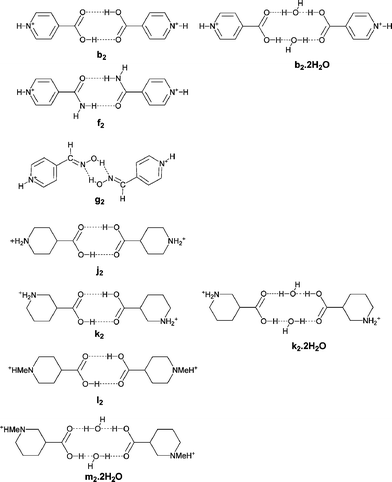
We have previously explored the use of supramolecular dications, in which two organic cations are linked by hydrogen bonds to form dimeric units.3h These supramolecular dications are potential structural mimics for molecular species such as 4,4′-bipyridinium ([4,4′-H2bipy]2+, a, see Scheme 1). For example, N-protonated isonicotinic acid (b, see Scheme 1) may form a supramolecular dication (b2, see Scheme 2) to mimic [4,4′-H2bipy]2+ through R2,2(8) dimerization (E) of the carboxylic acid functionality.3e,h
 | ||
| Scheme 1 Cationic tectons used. | ||
 | ||
| Scheme 2 Dimeric tectons. | ||
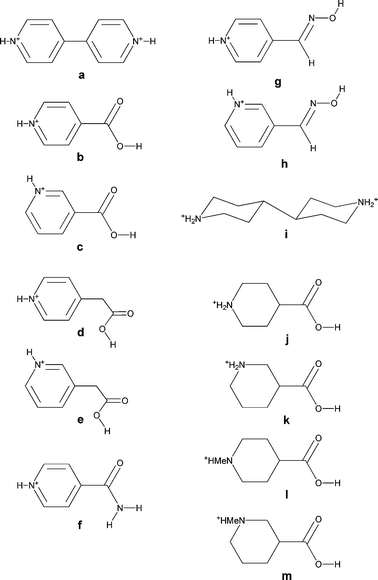
For the purposes of this study we have selected pyridinium and piperidinium derivatives bearing carboxylic acid, amide and oxime functionalities (see b–m, Scheme 1) because of their established tendency to form dimers (see ref. 12 and Scheme 2). Using cations based on piperidinium rather than pyridinium derivatives probes two principal effects: increasing the number of NH groups available and increasing the physical size of the organic cations from a relatively flat aromatic species to a bulky saturated group. The introduction of additional hydrogen-bond donors may lead to branching and increased dimensionality of the NH⋯anion hydrogen-bond network. We therefore sought to prepare [M(dto)2]2− salts of 4,4′-bipiperidinium, and protonated isonipecotic and nipecotic acids (j, k). We have also used the N-methylated analogues of the isonipecotic and nipecotic acids (l, m) in order to probe the effects of removing the equatorial NH hydrogen-bond donor sites in these species.
Cation variation allows assessment of a range of issues pertinent to creating diversity in the crystal structures synthesised:
(i) The viability of supramolecular dications such as those shown in Scheme 2.
(ii) The robustness of the inorganic/organic hybrid charge-assisted interactions (which are intended to link ions of opposite charge) and of the “organic” supramolecular synthons (which are intended to link cations) in the face of the competition offered by the other class of hydrogen bond. In these salts there is competition from hydrogen-bond donors other than H–N+ and between acceptors such as the carbonyl groups of the dto, carboxylic acid and amide groups.
(iii) The effects of changing the relative positions of the functional groups in isomeric cations (such as b and c, Scheme 1). This addresses a point we have previously raised—does the specific location of hydrogen-bond donor (or acceptor) groups matter in “engineering” the crystal structures of salts such as these?3f
(iv) The effect of increasing the separation and orientation of the functional groups (compare b and d, Scheme 1). This may offer further detailed control over the dimensions and geometry of the crystal structure.
Results and discussion
Salts 1–20, except 9, were prepared from the reaction of K2[M(dto)2] (M = Ni, Cu or Pt) and the hydrochloride salts of the relevant cation in aqueous solution. Salt 9 was prepared from acetonitrile solution from the reaction of the bis(benzyltriphenylphosphonium) salt of [Ni(dto)2]2− with the tetrafluoroborate salt of protonated isonicotinamide. Single crystals of 1–20 were prepared by a variety of crystallisation methods depending on the solubility of the products (see Experimental section for details). In each case the single-crystal structure was determined and the powder diffraction patterns recorded in order to confirm that the bulk phase corresponded to the single-crystal sample (which they did). The intramolecular geometry of the ions and molecules present in these structures are in general unremarkable and are not discussed. Summary crystallographic data are given in Tables 1 and 2 for 1–20. Hydrogen bonding details for 1–20 are given in Table 3. The syntheses and structures of salts 1, 3, 4, 13 and 19 have been previously reported10 and some details of their structures are rehearsed here for ease of reference.| 1 a | 2 | 3 a | 4 a | 5 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Chemical formula | C14H10N2O4S4Ni | C14H10N2O4S4Pt | C16H16N2O10S4Ni | C16H12N2O8S4Ni | C16H12N2O6S4Pt | C16H12N2O6S4Pt | C18H16N2O8S4Ni | C18H16N2O8S4Ni | C16H14N4O6S4Ni | C16H14N4O6S4Ni |
| a See ref. 10 for full details. | ||||||||||
| Crystal system | Triclinic | Triclinic | Monoclinic | Triclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group (no.) |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2) (2) |
P (2) |
P21/c (14) |
P (2) |
P (2) |
C2/c (15) | P21/n (14) | C2/c (15) |
P (2) |
P (2) |
| a/Å | 7.0246(7) | 7.1890(9) | 8.8334(28) | 7.0457(10) | 7.7014(12) | 17.464(3) | 4.0075(5) | 17.096(3) | 5.4904(11) | 7.3167(8) |
| b/Å | 8.1058(7) | 8.1431(11) | 11.3300(19) | 7.5275(10) | 8.4815(14) | 10.8917(16) | 16.375(2) | 10.3947(15) | 8.0895(16) | 8.6127(9) |
| c/Å | 8.3198(8) | 8.4188(11) | 11.0454(33) | 10.1365(14) | 8.6820(14) | 13.571(2) | 16.191(2) | 13.305(2) | 11.822(2) | 9.6402(11) |
| α/° | 76.768(2) | 84.248(2) | 90 | 75.683(2) | 72.757(3) | 90 | 90 | 90 | 106.80(3) | 63.690(2) |
| β/° | 68.567(2) | 67.789(2) | 96.037(25) | 70.904(2) | 67.078(2) | 128.397(2) | 90.063(11) | 112.760(2) | 99.07(3) | 70.051(2) |
| γ/° | 64.8560(10) | 63.871(2) | 90 | 85.902(2) | 77.891(3) | 90 | 90 | 90 | 92.32(3) | 82.668(2) |
| V/Å3 | 397.66(6) | 408.34 | 1099.3(5) | 492.22(12) | 494.22(14) | 2023.1(5) | 1062.5(2) | 2180.3(6) | 494.30(17) | 511.69(10) |
| T/K | 150(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) | 150(2) | 173(2) | 100(2) | 173(2) |
| Z | 1 | 1 | 2 | 1 | 1 | 4 | 2 | 4 | 1 | 1 |
| µ/mm−1 | 1.769 | 9.126 | 1.320 | 1.460 | 7.571 | 7.398 | 1.358 | 1.324 | 1.449 | 1.400 |
| Reflections collected | 2876 | 4315 | 2531 | 2229 | 5231 | 6387 | 5547 | 6910 | 5639 | 5380 |
| Independent reflections | 2085 | 1857 | 2122 | 1924 | 2242 | 2309 | 1872 | 2483 | 2252 | 2324 |
| R int | 0.0266 | 0.0293 | 0.0308 | 0.0234 | 0.0524 | 0.1049 | 0.1794 | 0.0260 | 0.0306 | 0.0235 |
| R 1 | 0.0332 | 0.0239 | 0.0258 | 0.0274 | 0.0391 | 0.0564 | 0.0455 | 0.0308 | 0.0328 | 0.0284 |
| 11 | 12 | 13 a | 14 | 15 | 16 | 17 | 18 | 19 a | 20 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Chemical formula | C16H14N4O6S4Ni | C14H22N2O4S4Ni | C16H24N2O8S4Ni | C16H24N2O8S4Pt | C16H24N2O8S4Cu | C16H28N2O10S4Ni | C16H28N2O10S4Pt | C16H28N2O10S4Cu | C16H28N2O8S4Ni | C18H28N2O8S4Ni |
| a See ref. 10 for full details. | ||||||||||
| Crystal system | Monoclinic | Triclinic | Triclinic | Triclinic | Monoclinic | Triclinic | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group (no.) | C2/c (15) |
P (2) |
P (2) |
P (2) |
C2/c (15) |
P (2) |
P (2) |
P (2) |
P21/c (14) | P21/c (14) |
| a/Å | 17.656(2) | 7.6921(4) | 7.5505(10) | 7.8728(9) | 23.276(4) | 6.1262(9) | 6.2542(11) | 6.1177(8) | 13.0682(8) | 8.1196(12) |
| b/Å | 10.0009(14) | 7.7577(4) | 7.9751(10) | 8.7532(10) | 9.1837(16) | 8.4035(13) | 8.4075(15) | 8.487(2) | 17.8121(10) | 12.1225(18) |
| c/Å | 13.3630(18) | 8.6131(4) | 9.9141(13) | 9.2079(10) | 11.898(2) | 12.7833(19) | 12.758(2) | 12.776(3) | 11.5482(7) | 13.528(2) |
| α/° | 90 | 100.562(3) | 92.959(2) | 79.481(2) | 90 | 104.596(2) | 104.227(3) | 104.937(18) | 90 | 90 |
| β/° | 118.784(2) | 99.931(3) | 104.109(2) | 77.692(2) | 117.028(3) | 95.445(2) | 95.401(3) | 95.186(14) | 110.414(1)) | 94.684(3) |
| γ/° | 90 | 103.836(3) | 103.856(13) | 65.701(2) | 90 | 106.733(2) | 107.964(3) | 106.614(14) | 90 | 90 |
| V/Å3 | 2068.0(5) | 475.99(4) | 558.28(2) | 562.27(11) | 2265.4(7) | 599.86(16) | 607.97(18) | 604.4(2) | 2519.3(3) | 1327(3) |
| T/K | 173(2) | 100(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) |
| Z | 4 | 1 | 1 | 1 | 4 | 1 | 1 | 1 | 4 | 2 |
| µ/mm−1 | 1.386 | 5.772 | 1.289 | 6.656 | 1.654 | 1.211 | 6.167 | 1.303 | 1.147 | 1.088 |
| Reflections collected | 10482 | 3714 | 5846 | 5876 | 9561 | 6305 | 6386 | 6263 | 15912 | 8357 |
| Independent reflections | 2372 | 1579 | 2533 | 2536 | 1992 | 2722 | 2755 | 2717 | 5763 | 3043 |
| R int | 0.0294 | 0.0340 | 0.0189 | 0.0259 | 0.1125 | 0.0322 | 0.0377 | 0.0257 | 0.0384 | 0.0427 |
| R 1 | 0.0261 | 0.0320 | 0.0263 | 0.0180 | 0.0448 | 0.0321 | 0.0299 | 0.0262 | 0.0326 | 0.0369 |
| dA⋯H/Å | dD⋯A/Å | ∠D–H⋯A/° | ∠D–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(dto)/° C(dto)/° |
||
|---|---|---|---|---|---|
| 1 | N–H⋯O | 1.95 | 152 | 118 | |
| 2.29 | 127 | 107 | |||
| 2 | N–H⋯O | 1.96 | 2.76 | 154 | 119 |
| 2.31 | 2.92 | 128 | 122 | ||
| 3 | N–H⋯O | 2.21 | 137 | 109 | |
| 2.02 | 140 | 140 | |||
| O–H⋯O | 2.23 | 113 | |||
| 1.69 | 160 | ||||
| 1.97 | 166 | ||||
| 4 | N–H⋯O | 2.15 | 138 | 115 | |
| 2.07 | 144 | 112 | |||
| O–H⋯O | 1.91 | 157 | |||
| 5 | N–H⋯O | 2.21 | 2.81 | 129 | 111 |
| 2.02 | 2.78 | 151 | 117 | ||
| O–H⋯O (dimer) | 1.80 | 2.62 | 164 | ||
| 6 | N–H⋯O | 2.02 | 2.81 | 149 | 119 |
| 2.18 | 2.85 | 134 | 113 | ||
| O–H⋯O (carb–dto) | 1.84 | 2.61 | 152 | ||
| 7 | N–H⋯O | 1.87 | 2.70 | 156 | 127 |
| 2.60 | 3.10 | 117 | 103 | ||
| O–H⋯O (carb) | 1.80 | 2.68 | 167 | ||
| 8 | N–H⋯O | 1.84 | 2.73 | 153 | 121 |
| 2.36 | 3.02 | 126 | 104 | ||
| O–H⋯O (carb) | 1.77 | 2.74 | 178 | ||
| O–H⋯S (carb) | 2.97 | 3.58 | 122 | ||
| 9 | N–H⋯O | 2.13 | 2.86 | 139 | 111 |
| 2.04 | 2.77 | 140 | 113 | ||
| N–H⋯O (amine) | 2.63 | 3.19 | 123 | ||
| N–H⋯S (amine) | 2.82 | 3.69 | 168 | ||
| N–H⋯O (dimer) | 2.04 | 2.91 | 174 | ||
| 10 | N–H⋯O | 1.99 | 2.77 | 148 | 121 |
| 2.40 | 3.00 | 126 | 107 | ||
| O–H⋯O (ox–dto) | 1.95 | 2.73 | 171 | ||
| N–H⋯O (py–ox) | 2.70 | 3.16 | 114 | ||
| 11 | N–H⋯O | 2.07 | 2.818 | 145 | 115 |
| 2.17 | 2.862 | 135 | 111 | ||
| O–H⋯O (ox–dto) | 1.88 | 2.72 | 175 | ||
| O–H⋯S (ox–dto) | 2.97 | 3.51 | 124 | ||
| N–H⋯O (py–ox) | 2.70 | 3.12 | 110 | ||
| 12 | N–H⋯O | 1.97 | 2.81 | 153 | 119 |
| 2.32 | 2.92 | 124 | 108 | ||
| 2.07 | 2.92 | 153 | 130 | ||
| 13 | N–H⋯O | 2.27 | 124 | 106 | |
| 1.88 | 156 | 118 | |||
| 2.04 | 157 | 134 | |||
| O–H⋯O | 1.89 | 174 | |||
| 1.71 | 168 | ||||
| 14 | N–H⋯O | 2.20 | 2.89 | 131 | 113 |
| 2.08 | 2.92 | 152 | 116 | ||
| O–H⋯O (dimer) | 1.76 | 2.59 | 169 | ||
| N–H⋯S | 2.88 | 3.65 | 151 | ||
| N–H⋯Pt | 2.71 | 3.52 | 160 | ||
| 15 | N–H⋯O | 2.06 | 2.82 | 139 | 113 |
| 2.13 | 2.89 | 140 | 113 | ||
| 2.01 | 2.87 | 155 | 124 | ||
| (COOH dimer, with H atoms disordered) | 1.81 | 2.64 | 176 | ||
| O–H⋯O | 1.79 | 2.61 | 167 | ||
| 16 | N–H⋯O | 2.00 | 2.90 | 163 | 119 |
| 2.34 | 2.85 | 114 | 108 | ||
| 1.83 | 2.81 | 176 | 122 | ||
| O–H⋯O (dto–H2O) | 2.14 | 2.90 | 156 | 115 | |
| O–H⋯O (COOH dimer) | 2.07 | 2.82 | 160 | ||
| 1.84 | 2.60 | 165 | |||
| 17 | N–H⋯O | 2.35 | 2.85 | 114 | 110 |
| 2.02 | 2.84 | 157 | 120 | ||
| 1.90 | 2.82 | 177 | 122 | ||
| O–H⋯O (dto–H2O) | 2.36 | 2.91 | 134 | 124 | |
| O–H⋯O (COOH–H2O) | 1.84 | 2.61 | 171 | ||
| 1.78 | 2.81 | 161 | |||
| 18 | N–H⋯O | 2.02 | 2.91 | 162 | 119 |
| 2.32 | 2.85 | 115 | 110 | ||
| 1.89 | 2.81 | 174 | 120 | ||
| O–H⋯O (dto–H2O) | 2.13 | 2.93 | 161 | 113 | |
| O–H⋯O (COOH dimer–H2O) | 2.14 | 2.81 | 158 | ||
| 19 | N–H⋯O | 2.21 | 138 | 112 | |
| 2.10 | 146 | 116 | |||
| O–H⋯O | 1.80 | 174 | |||
| 1.98 | 175 | ||||
| 20 | N–H⋯O | 2.12 | 2.89 | 141 | 117 |
| 2.26 | 2.94 | 133 | 107 | ||
| O–H⋯O (dto) | 2.14 | 2.87 | 169 | 113 | |
| O–H⋯O (COOH dimer with H atoms disordered) | 2.00 | 2.59 | 175 | ||
| 1.47 | 2.59 | 160 | |||
| 2.11 | 2.84 | 156 |
Pyridinium salts
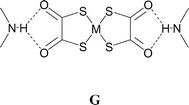
The salts [4,4′-bipyridinium][M(dto)2] [M = Ni (1) and Pt (2)] are formed in bulk as highly insoluble powders. Single crystals were therefore grown from the reaction mixtures by slow diffusion methods (see Experimental section). The structure of the platinum salt 2 (see Fig. 2) contains a nearly planar hydrogen-bonded ribbon of type F analogous to that seen in 1, through the formation of synthon D at either end of the [Pt(dto)2]2− dianion to give motif G.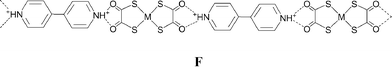
 | ||
| Fig. 2 The hydrogen-bonded ribbon motif in 2. | ||
The Pt⋯Pt separation in the ribbon is 20.33 Å, similar to the Ni⋯Ni separation in 1 (20.10 Å). The packing of the ribbons has a form seen in a range of pyridinium [PtCl4]2− salts,3 in which each NH hydrogen-bonded ribbon has six close-packed nearest neighbour ribbons (see Fig. 3). A similar ribbon packing is present in 1, and although the unit-cell dimensions of 1 and 2 are apparently different (see Table 1) the two are in fact isostructural. This is clear from an alternative unit cell assignment of the triclinic lattice of 2 which has metrics (a = 7.189, b = 8.150, c = 8.419 Å, α = 76.51, β = 67.79, γ = 63.77°) very close to those of 1 (see Table 1).
 | ||
| Fig. 3 Packing of the ribbons in 2. | ||
Single crystals of the isonicotinic and nicotinic acid derived salts [HNC5H4CO2H-4]2[Ni(dto)2][H2O]2 (3), [HNC5H4CO2H-3]2[Ni(dto)2] (4), [HNC5H4CO2H-4]2[Pt(dto)2] (5) and [HNC5H4CO2H-3]2[Pt(dto)2] (6), were synthesised through slow diffusion methods and their crystal structures determined (see Tables 1–3).
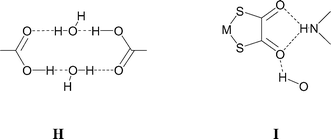
All four of these salts show formation of the synthon D at both ends of the anion giving rise to motif G. In the case of 3 a supramolecular dication is formed (b2·H2O, see Scheme 2) in which two molecules of water are incorporated between the carboxylic acid groups, and a cyclic R4,4(12) motif (H) results rather than E. The dication b2·H2O in 3 forms synthon D at both ends and a nearly planar hydrogen-bonded ribbon analogous to F results (see Fig. 4). Motif H also occurs in the analogous [PtCl4]2− salt3e and offers additional hydrogen-bond donor capability through the exocyclic OH groups. In 3 each of these OH groups forms a hydrogen bond of form I to a carbonyl functionality of a dto ligand in a neighbouring ribbon. This interaction is close to the plane of the dto ligand, approximately in the direction of the formally sp2 lone pair on the dto carbonyl group (see Table 2). The ribbon packing is of the six-nearest neighbour type but in this case there is significant (OH⋯O) hydrogen bonding (see Fig. 5). This leads to formation of a three-dimensional hydrogen-bond network between ions of CdSO4 (658) topology in which each anion is hydrogen bonded to four cations and vice versa.13
 | ||
| Fig. 4 The hydrogen-bonded ribbon motif in 3. | ||
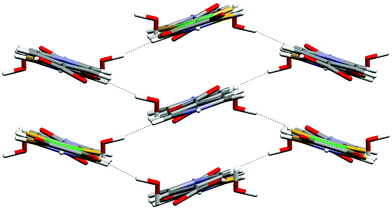 | ||
| Fig. 5 Packing of the ribbons in 3. | ||
In the analogous platinum salt [HNC5H4CO2H-4]2[Pt(dto)2] (5) there is no water incorporation in the supramolecular dication b2 and a near planar hydrogen-bond ribbon analogous to F results (see Fig. 6) with b2 effectively replacing its molecular analogue a (see Schemes 1 and 2). The control that such a formal substitution offers over the metrics of the structure is notable, as the Pt⋯Pt separation along the ribbon in 5 increases to 25.5 Å from 20.33 Å in 2. Once again the ribbons close pack in a quasi-hexagonal fashion.
 | ||
| Fig. 6 The hydrogen-bonded ribbon motif in 5. | ||
The protonated nicotinic acid salts [HNC5H4CO2H-3]2[Ni(dto)2] (4)10 and [HNC5H4CO2H-3]2[Pt(dto)2] (6) do not contain supramolecular dications. While similar, their structures are not identical. In both cases they may be interpreted by considering the packing of neutral supermolecules {[HNC5H4CO2H-3]2[M(dto)2]} (J) which are hydrogen bonded through each carboxylic acid group to a dto carbonyl group on a neighbouring supermolecule to form motif I, but the packing that results is different in 4 and 6. In 4 zigzag hydrogen-bonded ribbons are formed which pack side by side to give a sheet motif without OH⋯O hydrogen bonds between ribbons (see Fig. 7).
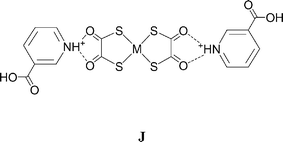
 | ||
| Fig. 7 The packing of the zigzag hydrogen-bonded ribbons in the sheet motif in 4. | ||
In 6 the carboxylic acid group is rotated by 180° relative to its orientation in 4, and a hydrogen-bonded sheet is formed (see Fig. 8). Salts 4 and 6 are not polymorphs but reveal two possible packings for essentially the same supermolecule (differing only in the metal at the centre of the anion and in the conformation of the carboxylate group). We have previously noted the possibility of “revealed” or “latent” polymorphism of this sort.3h This behaviour is not strictly (conformational) polymorphism,14 in that 4 and 6 are do not contain identical molecules, and so it might be better termed non-isomorphism! In any event it would be very surprising if the structure adopted by 6 were not accessible to 4 (and vice versa).
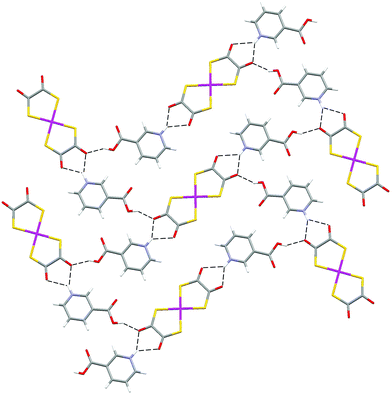 | ||
| Fig. 8 The hydrogen-bonded sheet motif in 6. | ||
The 4- and 3-pyridiniumacetic acid salts [HNC5H4CH2CO2H-4]2[Ni(dto)2] (7) and [HNC5H4CH2CO2H-3]2[Ni(dto)2] (8) again contain motif G. Notably, the NH⋯O distances in 7 are highly asymmetric (see Table 2) and synthon D is unusually distorted from planarity, with the plane of the pyridinium ring rotated by approximately 48° relative to the dto plane. As in 4 and 6 the carboxylic acid dimer E is not formed in either 7 or 8. Their structures may also be understood by considering the packing of neutral supermolecules, here of composition {[HNC5H4CH2CO2H]2[Ni(dto)2]} (analogous to J, see Fig. 9 and 10). As in 4 and 6 the supermolecules are linked by hydrogen-bond motifs of form I between carboxylic acid groups and dto carbonyls, close to the plane of the anion.
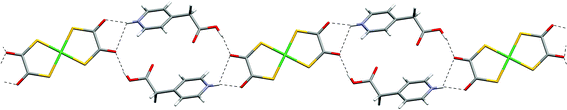 | ||
| Fig. 9 The hydrogen-bond ribbon motif in 7. | ||
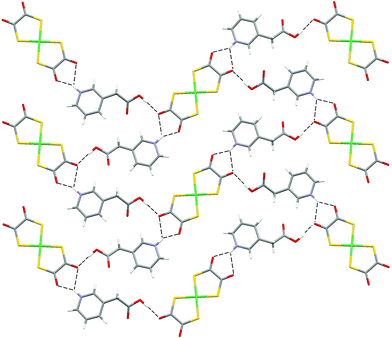 | ||
| Fig. 10 The hydrogen-bonded sheet motif in 8. | ||
In 7 zigzag ribbons are formed, which then pack so as to have six nearest neighbour ribbons. In contrast, the packing of the rather more planar supermolecules in 8 resembles that in 6 and a hydrogen-bonded sheet results.
[HNC5H4CONH2-4]2[Ni(dto)2] (9) was synthesised by the reaction of [BzPh3P]2[Ni(dto)2] with [HNC5H4CONH2-4][BF4] in acetonitrile, in order to avoid hydrolysis of the amide by aqueous acid. During the course of this synthesis, single crystals of [BzPh3P]2[Ni(dto)2] were prepared and characterised by X-ray methods (CCDC reference number 602658).
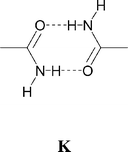
Single crystals of 9 were prepared through slow diffusion methods due to the insolubility (in MeCN) of the product. The anticipated supramolecular dication f2 (Scheme 2) is observed through formation of amide dimer interactions of type K. The dication forms synthon D at both ends and likewise the anion forms motif G. Therefore ribbons analogous to F result (see Fig. 11). As noted in the analogous [PtCl4]2− salt,3e the amide dimer has an exocyclic NH hydrogen-bond donor group that is able to cross-link ribbons. Such cross-linking occurs in 9 and hydrogen-bonded sheets are formed (see Fig. 11) as they do in [HNC5H4CONH2-4]2[PtCl4].3e However the sheets in 9 are somewhat pleated (whereas the sheets are nearly flat in the analogous [PtCl4]2− salt) and the amide dimer unit is rotated approximately 32.7° from the plane of the pyridinium ring (see Fig. 12). This is apparently associated with the formation of NH⋯S hydrogen bonds (together with a weaker NH⋯O bond) rather than the simple NH⋯O bond anticipated by analogy with [HNC5H4CONH2-4]2[PtCl4] (see Table 2; NH⋯S 2.82 Å, and N–H⋯S 168° cf. the N–H⋯O distance 2.63 Å and N–H⋯O angle 123°).
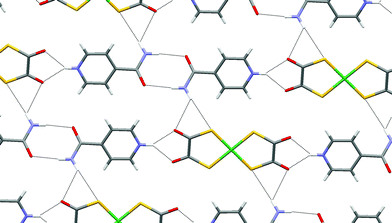 | ||
| Fig. 11 The hydrogen-bonded sheet motif in 9. | ||
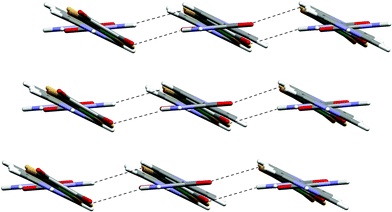 | ||
| Fig. 12 Packing of the sheet motif in the structure of 9. | ||
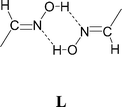
Single crystals of salts [HNC5H4CHNOH-4]2[Ni(dto)2] (10) and [HNC5H4CHNOH-3]2[Ni(dto)2] (11) of the 4- and 3-pyridiniumoxime cations were prepared by slow diffusion methods. They both contain motif G but the known12,15 oxime dimer L is not formed in either case. Their structures may be viewed as consisting of the packing of approximately planar neutral supermolecules of composition {[HNC5H4CHNOH]2[Ni(dto)2]} (analogous to J, see Fig. 13 and 14). As in 4 and 6 (and 7 and 8) the supermolecules are linked by hydrogen-bond motifs of form I, here between oxime hydroxyl groups and dto carbonyls, close to the plane of the anion. In 10 this leads to the formation of a zigzag ribbon motif (as in 4 and 7), shown in Fig. 13, and as in 4 these ribbons are essentially planar and pack side-by-side to form sheets.
 | ||
| Fig. 13 The sheet formed by zigzag hydrogen-bonded ribbons in 10. | ||
 | ||
| Fig. 14 The hydrogen-bonded sheet in 11. | ||
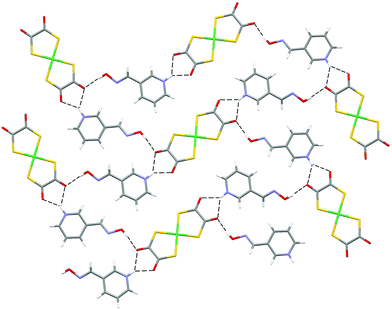
Salt 11 forms the alternative two dimensional hydrogen-bonded network motif analogous to those observed in 6 and 8 (see Fig. 14).
Piperidinium salts
Reaction of [4,4′-bipiperidinium]Cl2 with K2[Ni(dto)2] in aqueous solution gives salt [4,4′-H2bipip][Ni(dto)2] (12) as a highly insoluble purple solid, and single crystals for X-ray analysis were prepared using slow diffusion methods. Its structure contains synthon D formed by the equatorial NH on each piperidinium ring at both ends of each cation and anion, and a hydrogen-bonded ribbon of form F results (see Fig. 15). As in the analogous [PtCl4]2− salt,3e the axial NH group is able to form additional hydrogen bonds and a branched NH⋯anion network is formed in which each anion is hydrogen bonded to four cations and vice versa. In 12, as in [4,4′-H2bipip][PtCl4],3e the hydrogen-bond network is two dimensional and of the [4,4] type13 (see Fig. 15). The NH⋯O hydrogen bonds in 12 are all approximately in the plane of the anions and are topologically related to motif I, but here in a doubled form M.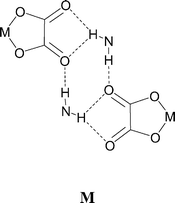
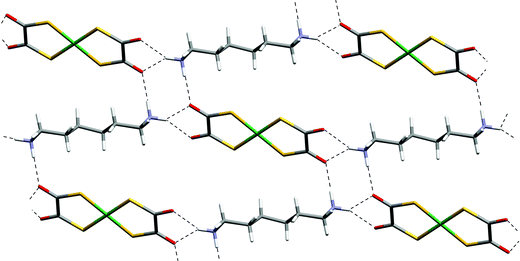 | ||
| Fig. 15 The hydrogen-bonded sheet motif in 12. | ||
The [Ni(dto)2]2− salt of protonated isonipecotic acid [H2NC5H9CO2H-4]2[Ni(dto)2] (13) contains the dication j2 (see Scheme 2), a supramolecular mimic of [4,4′-H2bipip]2+. Indeed the crystal structure of 13 resembles that of 12 in containing hydrogen-bonded sheets of similar form in respect of the NH⋯dto hydrogen bonding (see Fig. 16).
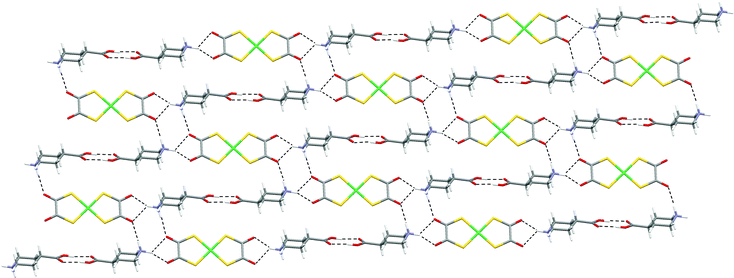 | ||
| Fig. 16 The hydrogen-bonded sheet motif in 13. | ||
The analogous platinum and copper salts [H2NC5H9CO2H-4]2[Pt(dto)2] (14) and [H2NC5H9CO2H-4]2[Cu(dto)2] (15) were synthesised in an similar manner to 13, and single crystals suitable for X-ray diffraction study were prepared by evaporation in each case. Although all three salts share structural features, there are significant differences between their crystal structures.
Both 14 and 15 contain the supramolecular dication j2, and a hydrogen-bonded ribbon motif of type F results through participation of the equatorial piperidinium NH groups (see Fig. 17 and 18). In contrast to all other cases in this paper, in 14 the additional hydrogen-bond donors of the cation, the axial NH groups, bond to dto sulfur atoms in neighbouring ribbons. A two-dimensional [4,4] network of ions each linked to four neighbours by hydrogen bonds results (see Fig. 17).
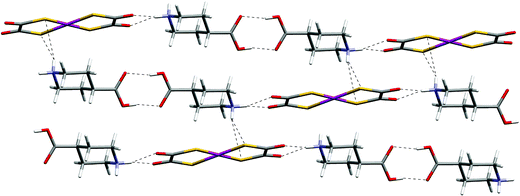 | ||
| Fig. 17 The NH⋯O and NH⋯S hydrogen-bonded sheet motif in 14. | ||
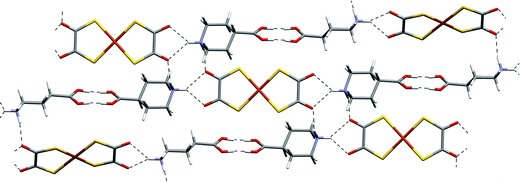 | ||
| Fig. 18 The hydrogen-bonded sheet in 15. | ||
The structure of the copper analogue, 15, is similar to that of 13 with a hydrogen-bonded sheet motif being formed (Fig. 18). The sheets pack in a similar but not identical manner to 12 and 13. Notably the [Cu(dto)2]2− anions in 15 are non-planar (the copper coordination plane is 18.3° from the plane of the dto ligand).
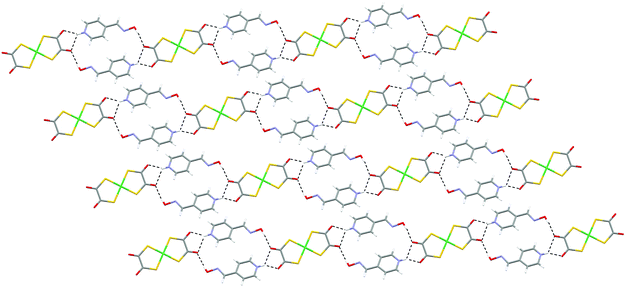
The three [M(dto)2]2− salts of protonated nipecotic acid [H2NC5H9CO2H-3]2[M(dto)2][H2O]216 (M = Ni), 17 (M = Pt) and 18 (M = Cu) are isostructural, crystallising in the space group P, with only small differences in the unit cell dimensions as a result of the variation in metal atom. In each case a centrosymmetric, hydrated supramolecular dication k2·H2O (Scheme 2) is present, which contains the R4,4(12) motif H as seen in 3. This species therefore has both the NH2 functionalities characteristic of piperidinium-based tectons and exocyclic OH hydrogen-bond donor groups. The dication k2·H2O is therefore unique among the dications in this paper in having a total of six OH or NH hydrogen-bond donor groups rather than the four present in f2, j2, k2, b2·H2O and i.
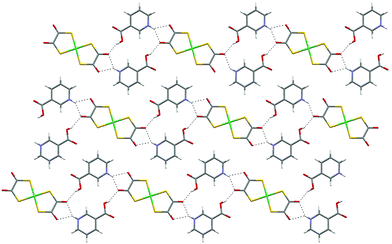
The supramolecular structure of 16–18 shows similarities to those of 12, 13 and 15, with synthon I found at both ends of the anion and likewise at both ends of the dication k2·H2O. However there are significant differences in detail and in the overall structure from those of 12, 13 and 15. In 16–18 the primary D-type hydrogen bonding involves the axial piperidinium NH donor rather than the equatorial. As a result the ribbons formed are highly non-planar (see Fig. 19). As a further corollary, the cross-linking of ribbons involves the equatorial piperidinium NH groups.
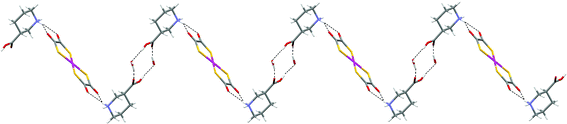 | ||
| Fig. 19 The zigzag hydrogen-bonded ribbon formed in the structures of 16–18. | ||
The zigzag ribbons are linked by both NH⋯dto carbonyl interactions and exocyclic water OH⋯dto carbonyl hydrogen bonds, in both cases these bonds lying close to the plane of the ligand. In total each anion accepts hydrogen bonds from six dications and each dication donates hydrogen bonds to six anions. The three dimensional network formed is rather open (see Fig. 20) and contains large pores which are filled with a second (identical) network interpenetrating but not strongly hydrogen bonding to the first (see Fig. 21). The networks are related by a unit cell translation along the a axis.
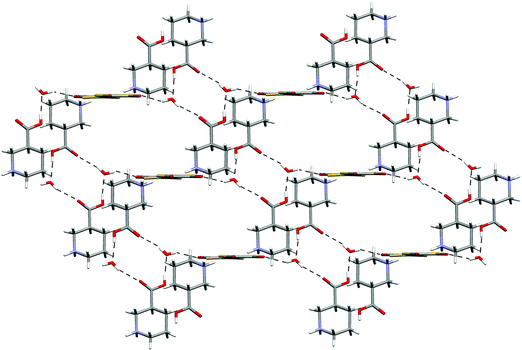 | ||
| Fig. 20 The open three-dimensional network in the structures of 16–18. | ||
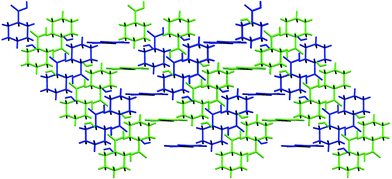 | ||
| Fig. 21 Interpenetration of the two identical networks in the structures of 16–18. | ||
Binding of axial piperidinium NH protons is also observed in the structure of [H(Me)NC5H9CO2H-4]2[Ni(dto)2] (19),10 where it is enforced through (equatorial) N-methylation of the isonipecotic acid prior to protonation to form cation l. In 19, this leads to the formation of a zigzag ribbon motif (see Fig. 22). Salt 19 contains a (non-hydrated) supramolecular dication l2 (see Scheme 2) which forms synthon D at both its ends. Formation of motif G leads to the zigzag ribbon in Fig. 22 but in the absence of any other good hydrogen-bond donors in the dication the hydrogen-bond network formed is perforce one dimensional. The ribbon formed is reminiscent of that in 16–18 in which, as in 19, the axial NH group forms the D-type interaction.
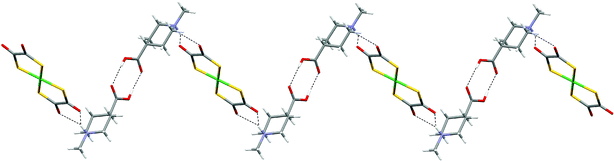 | ||
| Fig. 22 The zigzag hydrogen-bonded ribbon formed in the structure of 19. | ||
Salt [H(Me)NC5H9CO2H-3]2[Ni(dto)2][H2O]2 (20) is the methylated nipecotic acid-derived analogue of 19. There is a two site orientational disorder of the carboxylic acid groups in the crystal structure. This structure contains a hydrated supramolecular dication in which two cations m are linked through interaction H to afford m2·H2O (see Scheme 2). As expected, the methyl group occupies the equatorial position on the piperidinium leaving an axial NH group. A zigzag ribbon results with anion motif G formed. Cross-linking of ribbons occurs through the exocyclic OH donors to form a pleated hydrogen-bonded sheet motif, shown in Fig. 23.
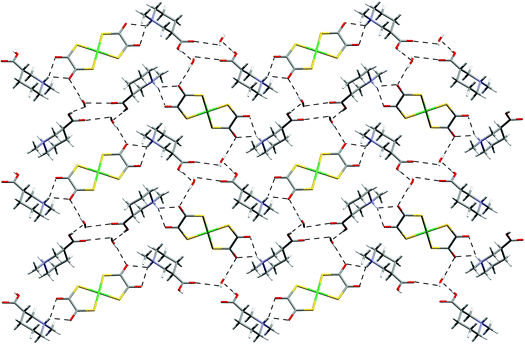 | ||
| Fig. 23 The sheet motif found in the structure of 20. Only one of the disordered carboxylic group orientations is shown for clarity. | ||
Conclusions
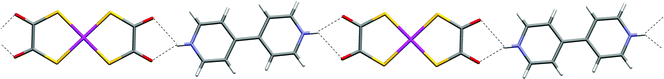
In crystal synthesis at its most basic level we seek control over the stoichiometry of the product crystals. Therefore inclusion of the desired ions is necessary and exclusion of the counterions or the solvent(s) used in the reactions is required. In the work reported herein we have seen occasional incorporation of water in the products (in 3, 16–18 and 20) but otherwise the synthetic strategy provides inherently good control over composition, as required by charge balance considerations. At a higher level we seek to form the desired synthon(s) and so the formation of the required supramolecular and periodic motifs and hence the entire crystal structure. This second level of control, over molecular recognition between tectons, requires that the supramolecular synthons be robust—i.e. they form. In the present work the NH⋯dto synthon of type D forms in all cases, showing exceptional robustness considering the wealth of alternative hydrogen-bond acceptors present in all cases (including the thiocarboxylate acceptor present in all the anions). Indeed, motif G in persists in all pyridinium dithiooxalate structures in the Cambridge Structural Database (CSD)16 as well as in 1–20. Synthon D is apparently much more robust than the (non-charge assisted) “organic” synthons used here (such as the carboxylic acid dimer), which have a much lower rate of occurrence in our structures. The robustness of D does not imply that it is particularly rigid (see Fig. 24). In fact the NH⋯O hydrogen bonds vary in length and most of the interactions are typically rather asymmetric. Indeed it may be that flexibility of this sort is central to the robustness of the synthon-it is able to respond at low energy cost to the environment around it. Importantly synthon D may be readily generalised to 3- and 4-piperidinium derivatives as well as 3- and 4-pyridinium species. This is attractive since the piperidinium species afford branching of the hydrogen-bond networks through their NH2 groups.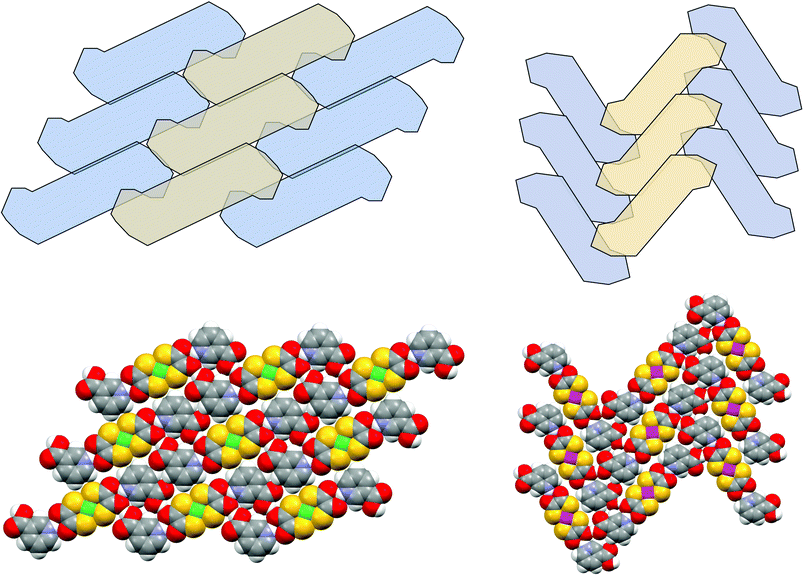 | ||
| Fig. 24 Supermolecule packing motifs (ii) (as observed in 4, 7 and 10), and (iii) (as in 6, 8, 11); space-filling diagrams of 4 and 6. | ||
The robustness of motif G allows structures of the form [pyridinium]2[M(dto)2] to be regarded as being formed by the packing of neutral supermolecules analogous to J. Thus in 4–11 the pyridinium substituents form hydrogen bonds to their neighbours and one of three alternative structure types results: type (i) in which linear chains are formed through R(2,2)8 dimerisation of the 4-CO2H and –CONH2 substituents (as in 5 and 9); type (ii) in which zigzag ribbons are arranged in layers without OH⋯O hydrogen bonds between ribbons (in 4, 7 and 10); type (iii) in which the supermolecules are linked by OH⋯O hydrogen bonds into a sheet (in 6, 8 and 11). In both type (ii) and (iii) structures the hydrogen bonds between J-type supermolecules are formed between donors not collinear with the axis of the pyridinium group and thiocarboxylates of neighbouring anions (see Fig. 24). In these structures the planned “organic” synthons do not form. Apparently the residual hydrogen-bond acceptor capability of the thiocarboxylate groups is too powerful to allow formation of carboxylic acid dimers (in 6, for example).
Whilst in this work we have noted cases of isomorphism (as in 16–18) and no cases of polymorphism, there is reason to believe that polymorphs of some structures are likely given that such similar tectons give rise to different structure types (e.g.4 and 6). We have termed this behaviour ‘latent polymorphism’, in the sense that possible polymorphs have been identified but have not yet been made.
The use of isomeric cations in this work allows direct comparison of packing efficiency in some of the salts (e.g.5 and 6; 7 and 8; 10 and 11). In all three cases the 4-substituted cations form structures with lower cell volumes per cation than do the 3-substituted cations (494 vs. 506; 531 vs. 545; and 512 vs. 517 Å3 for 5 and 6; 7 and 8; 10 and 11, respectively).
Finally we note that the model of supramolecular synthesis given here is clearly heuristic and explicitly ignores a range of issues (electrostatics; shape fit; all the weaker CH⋯O/S/M contacts; π–π effects etc.) that must contribute, probably rather significantly, to the stability of the structures observed.
Experimental
Synthesis
The synthesis of salts 1, 3, 4, 13 and 19 have been published previously.10 The salts K2[Ni(dto)2], K2[Cu(dto)2] and K2[Pt(dto)2] (dto = dithiooxalate) were prepared according to the published procedures.17 All pyridines, except N-methylnipecotic acid and 4- and 3-pyridylacetic acids, were purchased from Lancaster or Aldrich and recrystallised from concentrated hydrochloric acid prior to use. N-Methylnipecotic acid hydrochloride was prepared from nipecotic acid according to the published procedure.18 4- and 3-pyridylacetic acid and 4,4′-bipiperidine as its hydrochloride salt were purchased from either Lancaster or Aldrich as the hydrochloride salts. Elemental analyses were performed by the School of Chemistry Microanalytical Service. Powder diffraction was carried out using a Bruker D8 diffractometer. Details of yields, colours and elemental analysis for the new salts 2, 5–12, 14–18 and 20 are given in Table 4. All manipulations were carried out in air using solvents of reagent grade as supplied, without further purification or drying.| Elemental analysisa (%) | |||||
|---|---|---|---|---|---|
| Compound | Colour | Yield (%) | C | H | N |
| a Calculated values in parentheses. | |||||
| [4,4′-H2bipy][Pt(dto)2] 2 | Deep red | 76 | 27.99 (28.30) | 1.85 (1.70) | 4.50 (4.70) |
| [HNC5H4CO2H-4]2[Pt(dto)2] 5 | Deep red | 75 | 27.59 (28.11) | 1.56 (1.77) | 3.96 (4.10) |
| [HNC5H4CO2H-3]2[Pt(dto)2] 6 | Deep red | 86 | 27.66 (28.11) | 1.85 (1.77) | 3.89 (4.10) |
| [HNC5H4CH2CO2H-4]2[Ni(dto)2] 7 | Purple | 81 | 37.05 (37.58) | 2.44 (2.80) | 4.23 (4.87) |
| [HNC5H4CH2CO2H-3]2[Ni(dto)2] 8 | Purple | 74 | 37.51 (37.58) | 2.94 (2.80) | 4.90 (4.87) |
| [HNC5H4CONH2-4]2[Ni(dto)2] 9 | Purple | 83 | 35.33 (35.24) | 2.76 (2.59) | 10.28 (10.28) |
| [HNC5H4CHNOH-4]2[Ni(dto)2] 10 | Purple | 62 | 35.59 (35.24) | 2.47 (2.59) | 10.05 (10.28) |
| [HNC5H4CHNOH-3]2[Ni(dto)2] 11 | Purple | 79 | 35.45 (35.24) | 2.46 (2.59) | 10.21 (10.28) |
| [4,4′-H2bipip][Ni(dto)2] 12 | Purple | 73 | 35.43 (35.99) | 4.35 (4.310 | 5.57 (6.00) |
| [H2NC5H9CO2H-4]2[Pt(dto)2] 14 | Deep red | 66 | 27.36 (27.70) | 3.21 (3.20) | 3.69 (4.04) |
| [H2NC5H9CO2H-4]2[Cu(dto)2] 15 | Dark green | 46 | 34.50 (34.06) | 3.91 (4.29) | 4.83 (4.97) |
| [H2NC5H9CO2H-3]2[Ni(dto)2][H2O]216 | Purple | 60 | 32.31 (32.28) | 4.96 (4.74) | 4.73 (4.71) |
| [H2NC5H9CO2H-3]2[Pt(dto)2][H2O]217 | Deep red | 89 | 25.92 (26.26) | 3.93 (3.86) | 3.50 (3.83) |
| [H2NC5H9CO2H-3]2[Cu(dto)2][H2O]218 | Dark green | 36 | 31.86 (32.02) | 4.91 (4.70) | 4.38 (4.67) |
| [H(Me)NC5H9CO2H-4]2[Ni(dto)2][H2O]220 | Purple | 40 | 34.01 (34.68) | 5.36 (5.17) | 4.31 (4.49) |
Compounds 5, 6, 7, 8, 10, 11 and 12 were prepared by the same method from the potassium bis(dithiooxalate) salt of the appropriate metal and the hydrochloride salt of the appropriate base, and crystals suitable for X-ray diffraction grown in the same way.
Compounds 15, 17 and 18 were prepared by the same method from the potassium bis(dithiooxalate) salt of the appropriate metal and the appropriate hydrochloride.
Compound 20 was prepared by the same method from K2[Ni(dto)2] and N-methylnipecotic acid hydrochloride.
X-Ray diffraction
Diffraction measurements on 2, 5, 6, 8–11, 14–18 and 20 were made on Bruker-AXS three-circle CCD diffractometers using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Diffraction measurements for 7 were made using a Bruker SMART diffractometer on station 9.8 at the Daresbury SRS. Unit cell dimensions were determined from reflections taken from three sets of ten frames (at 0.3° steps in ω), each at 10 s exposure. Data frames were referenced to a set of ten dark frame readings each taken at n s exposures without X-rays, where n s is the time for each exposure during data collection. For crystals of monoclinic or higher crystal symmetry, a hemisphere of reciprocal space was scanned by 0.3 θ steps at φ = 0, 88 and 180° with the area detector centre held at 2θ = 27°. For triclinic crystals, a full sphere of reciprocal space was scanned. In all experiments, each exposure was n s (typically, 10 ≤ n ≤ 40). The reflections were integrated using the SAINT18 program. Absorption, Lorentz and polarisation corrections were applied. The structures were solved by direct methods and refined using full-matrix least squares against F2 using SHELXTL.19 All non-hydrogen atoms were assigned anisotropic displacement parameters and refined without positional constraints. Hydrogen atoms were included in idealised positions with isotropic displacement parameters constrained to 1.5 times the Uequiv of their attached carbon atoms for methyl hydrogens, and 1.2 times the Uequiv of their attached carbon atoms for all others.Diffraction measurements for [BzPh3P]2[Ni(dto)2] and 12 were made at −173 °C on a Bruker-AXS Proteum diffractometer equipped with a CCD area detector using Cu-Kα radiation (λ = 1.54178 Å). The X-ray beam was collimated and focused. Data were collected as a series of narrow frames covering 0.3° in ω or φ and integrated using the SAINT program.19 The structures were solved and refined as described above.
CCDC reference numbers 183068–183072 and 602658–602673.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b604319d
Powder X-ray measurements were performed using Cu-Kα radiation (λ = 1.5418 Å) on a Bruker-AXS D8-Advance diffractometer equipped with a secondary monochromator. The data were collected in the range 5 ≤ 2θ ≤ 60° in θ–θ mode with a step time of n s (5 < n < 10) and step width of 0.02°. Powder diffraction patterns of bulk samples of 2, 5–9, 10–12, 14–18 and 20 were consistent with them having the same structure as the single-crystal samples analysed (see ESI†).
Acknowledgements
We thank the University of Bristol and the EPSRC for financial support.References
- (a) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS; (b) G. R. Desiraju, Chem. Commun., 1997, 1475 RSC.
- (a) M. Simard, D. Su and J. D. Wuest, J. Am. Chem. Soc., 1991, 113, 4696 CrossRef; (b) J. H. Fournier, T. Maris, J. D. Wuest, W. Z. Guo and E. Galoppini, J. Am. Chem. Soc., 2003, 125, 1002 CrossRef CAS; (c) S. Mann, Nature, 1993, 365, 499 CrossRef CAS; (d) A. Jouaiti, M.-W. Hosseini and N. Kyritsakas, Chem. Commun., 2003, 472 RSC , and references cited therein.
- (a) G. R. Lewis and A. G. Orpen, Chem. Commun., 1998, 1873 RSC; (b) A. L. Gillon, A. G. Orpen, J. Starbuck, X.-M. Wang, Y. Rodríguez-Martín and C. Ruiz-Pérez, Chem. Commun., 1999, 2287 RSC; (c) A. L. Gillon, G. R. Lewis, A. G. Orpen, S. Rotter, J. Starbuck, X.-M. Wang, Y. Rodríguez-Martín and C. Ruiz-Pérez, J. Chem. Soc., Dalton Trans., 2000, 3897 RSC; (d) B. Dolling, A. L. Gillon, A. G. Orpen, J. Starbuck and X.-M. Wang, Chem. Commun., 2001, 567 RSC; (e) A. Angeloni and A. G. Orpen, Chem. Commun., 2001, 343 RSC; (f) A. Angeloni, P. C. Crawford, A. G. Orpen, T. J. Podesta and B. J. Shore, Chem. Eur. J., 2004, 10, 3783 CrossRef CAS; (g) T. J. Podesta and A. G. Orpen, Cryst. Growth Des., 2005, 5, 681 CrossRef CAS; (h) C. J. Adams, A. Angeloni, A. G. Orpen, T. J. Podesta and B. J. Shore, Cryst. Growth Des., 2006, 6, 411–422 CrossRef CAS.
- (a) J. C. M. Rivas and L. Brammer, Inorg. Chem., 1998, 37, 4756 CrossRef; (b) L. Brammer, J. K. Swearingen, E. A. Bruton and P. Sherwood, Proc. Natl. Acad. Sci. USA, 2002, 99, 4956 CrossRef CAS; (c) L. Brammer, G. M. Espallargas and H. Adams, CrystEngComm, 2003, 5, 343 RSC.
- P. C. Crawford, A. L. Gillon, J. Green, A. G. Orpen, T. J. Podesta and S. V. Pritchard, CrystEngComm, 2004, 6, 419 RSC.
- (a) S. Ferlay, V. Bulach, O. Felix, M.-W. Hosseini, J.-M. Planeix and N. Kyritsakas, CrystEngComm, 2002, 4, 447 RSC; (b) S. Ferlay, O. Felix, M.-W. Hosseini, J.-M. Planeix and N. Kyritsakas, Chem. Commun., 2002, 702 RSC; (c) S. Ferlay and M.-W. Hosseini, Chem. Commun., 2004, 788 RSC.
- (a) M. Felloni, P. Hubberstey, C. Wilson and M. Schröder, CrystEngComm, 2004, 6, 87 RSC; (b) K. Kumar, A. Ballabh, D. A. Jose, P. Dastidar and A. Das, Cryst. Growth Des., 2005, 5, 651 CrossRef; (c) X. Ren, J. Xie, Y. Chen and R. K. Kremer, J. Mol. Struct., 2003, 660, 139 CrossRef CAS; (d) M. Fourmigué, C. Mézière and S. Dolou, Cryst. Growth Des., 2003, 3, 806; (e) J. Valdés-Martínez, M. Del Rio-Ramirez, S. Hernández-Ortega, C. B. Aakeröy and B. Helfrich, Cryst. Growth Des., 2001, 1, 485 CrossRef CAS.
- (a) C. B. Aakeröy, K. Beffert, J. Desper and E. Elisabeth, Cryst. Growth Des., 2003, 34, 837 CrossRef; (b) V. Bertolasi, L. Pretto, P. Gilli, V. Ferretti and G. Gilli, New J. Chem., 2002, 26, 1559 RSC; (c) K. T. Holman, A. M. Pivovar, J. A. Swift and M. D. Ward, Acc. Chem. Res., 2001, 34, 107 CrossRef CAS.
- D. Braga, L. Maini, M. Polito, E. Tagliavini and F. Grepioni, Coord. Chem. Rev., 2003, 246, 53 CrossRef CAS.
- T. J. Podesta and A. G. Orpen, CrystEngComm, 2002, 4, 336 RSC.
- W. Dietzsch, P. Strauch and E. Hoyer, Coord. Chem. Rev., 1992, 121, 43 CrossRef CAS.
- F. H. Allen, W. D. S. Motherwell, P. R. Raithby, G. P. Shields and R. Taylor, New J. Chem., 1999, 23, 25 RSC.
- (a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1461 CrossRef; (b) M. O'Keeffe, M. Eddaoudi, H. L. Li, T. Reineke and O. M. Yaghi, J. Solid State Chem., 2000, 152, 3 CrossRef CAS.
- (a) J. Bernstein and A. T. Hagler, J. Am. Chem. Soc., 1978, 100, 673 CrossRef CAS; (b) A. T. Hagler and J. Bernstein, J. Am. Chem. Soc., 1978, 100, 6349 CrossRef CAS; (c) I. Bar and J. Bernstein, J. Phys. Chem., 1982, 86, 3223 CrossRef; (d) I. Bar and J. Bernstein, J. Phys. Chem., 1984, 88, 243 CrossRef CAS.
- C. B. Aakeröy, A. M. Beatty and D. S. Leinen, CrystEngComm, 2002, 4, 310 RSC.
- (a) F. H. Allen, J. E. Davies, J. J. Galloy, O. Johnson, O. Kennard, C. F. Macrae, E. M. Mitchell, G. F. Mitchell, J. M. Smith and D. G. Watson, J. Chem. Inf. Comput. Sci., 1987, 31, 187; (b) F. H. Allen and O. Kennard, Chem. Des. Autom. News, 1993, 8, 1 & 31 Search PubMed.
- (a) E. G. Cox, W. Wardlaw and K. C. Webster, J. Chem. Soc., 1935, 1475 RSC; (b) R. G. Pearson and D. A. Sweigart, Inorg. Chem., 1970, 9, 1167 CrossRef CAS.
- A. P. Gray, R. D. Platz, T. R. Henderson, T. C. P. Chang, K. Takahashi and K. L. Dretchent, J. Med. Chem., 1988, 31, 807 CrossRef CAS.
- SMART, Copyright 1989–1999, Bruker AXS, Madison, WI, USA; SAINT, Bruker AXS, Madison, WI, USA; SHELXTL, Rev. 5.0, Bruker AXS, Madison, WI, USA.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and calculated powder XRD spectra. See DOI: 10.1039/b604319d |
| This journal is © The Royal Society of Chemistry 2006 |