Effect of methylation on the coordination of copper by small azacryptands; the role of geometrically constrained hydrogen bonding in stabilizing terminally coordinated oxygen species†
David
Farrell
a,
Charles J.
Harding
c,
Vickie
McKee
b and
Jane
Nelson
b
aDept. of Chemistry Queens University Belfast, UK BT9 5AG
bDept. of Chemistry Loughborough University, Leics., UK LE11 3TU
cDept. of Chemistry, Open University, Milton Keynes, UK MK7 6AA
First published on 10th April 2006
Abstract
Treatment of N-methyl substituted aminocryptand hosts with copper(II) generates monocopper(II) cryptates where copper(II) coordinates an oxygen-centered species, formally H3O+, which is also strongly hydrogen bonded to three aminocryptand N-methyl atoms via bonds which may best be viewed as NHδ+⋯Oδ− in consequence of charge transfer. The strength of this hydrogen bonding precludes successful competition of another copper ion for the second coordination site thus suppressing formation of any Cu–Cu bonded average-valent system.
Introduction
The relatively small cryptand (L1), allied to Sargeson’s N-bridgehead cryptand1sep has shown itself to be a useful complexant of cations from the transition2–4 or main group3,5 series, as well as, in its protonated state, for the smallest anions6–8 fluoride or chloride. The 2-carbon separation of the N atoms in the cap protects the ligand from the reactivity associated with the gem-diamino assembly, which has limited the usefulness of sep in comparison with the carbon-bridgehead analogue sar. The properties of the small azacryptand host L1 include enforcement of copper–copper bonding in the unusual average-valence +1.5 redox state, a state of considerable interest in biology, where several important multielectron redox sites (the CuA site of several cytochrome c oxidases9 and nitrous oxide reductases10) make use of the easy electron transfer it enables.The average valence dicopper (1.5) redox state has been shown to occur in the azacryptand series in three situations where the host incorporates a 2-C link between tetraamino caps, i.e. in L0 and L4 as well as in L1. We wished to discover whether the range of small azacryptand hosts which mimic this unusual biosite could be extended, and, in a first attempt at functionalisation, have synthesised methylated versions of L1 in order to investigate their complexation properties.
Results and discussion
Our initial attempt at methylation of L1, using MeI as alkylating agent, resulted in the generation of the tetramethyl cryptand L2 in its diprotonated state. On the other hand, the use of the formic acid/formaldehyde reaction mixture, earlier used for this purpose by Lehn11 and coworkers, is known to generate the perN-methyl cryptand, L3. It is interesting that different methylation strategies achieve such different outcomes; this may partly be due to pH effects, in that the first step in the formic acid/formaldehyde route is facilitated by protonation is not involved in the nucleophilic attack of the amine lone pair on MeI in the second reaction. Solubility factors must also play their part, as the solvents in the two reactions are very different. The difference in methylation level consequent on these two different strategies is potentially useful in that it could allow us to monitor any gradation in properties resulting from substitution of NH protons by the bulkier N-methyl groups.In macrocyclic ligands, permethylation of the sec-amino function has been shown12 to have dual consequences: an increase in the size of the host, and a decrease in its basicity. In cryptands, the distortion cannot always be described as a general expansion of the host cavity: when NH functions in sar are methylated13 to generate the Me6sar host, for example, the geometric consequence of repulsion between the methyl groups is to destabilize the threefold symmetry of the complex, leaving two N-donors uncoordinated and causing a coordinating ion such as Ni(II) to revert to four-coordination in what becomes essentially a strapped macrocyclic arrangement. In cryptands with a pair of ready-made tetraamine coordination sites, another response to the steric requirements of N-substitution could be to use one end of the host for cation coordination, allowing relaxation of constraint to occur at the other end. In order to evaluate the prospects for generation of functionalised average-valence cryptands, we wished to investigate the response adopted by the relatively flexible substituted-L1 skeleton.
Tetramethylated system
Use of the MeI/K2CO3 methylating system generates a white solid whose infrared spectrum testifies to the presence of both NH and NMe functions. Qualitative tests with aqueous AgNO3 indicate the presence of iodide. Treatment of the iodide salt of the cryptate, (L2H2I2·2Me2CO) 1, with two equivalents of copper(II) salt in ethanol generates Cu(II) products. Recrystallisation of the product of reaction of this salt with copper nitrate, from DMF, gave blue–green crystals on THF diffusion, which did not show the intense electronic absorption characteristic2 of average-valence dicopper although analyzing for three nitrate ions, suggesting a tripositively charged cation. The crystals, which proved to be [CuL2(H3O)](NO3)3·MeCN, 2a, were suitable for X-ray crystallography and the structure of the cation is shown in Fig. 1.![Structure of the [CuL2(H3O)]3+ cation of cryptate 2a. Hydrogen bonds shown as solid lines and the minor component of the disorder has been omitted for clarity.](/image/article/2006/DT/b602003h/b602003h-f1.gif) | ||
| Fig. 1 Structure of the [CuL2(H3O)]3+ cation of cryptate 2a. Hydrogen bonds shown as solid lines and the minor component of the disorder has been omitted for clarity. | ||
The explanation for the observations above is obvious, when it is realized that the second encapsulated atom responsible for the tripositive-cationic formulation is not another copper ion but is (formally) H3O+, in the cation, [CuL2(H3O)]3+. There is 66% : 33% disorder in occupancy of the encapsulated (Cu, O) sites. In the major component of the disorder, N⋯O contacts from N4A, N4B and N4C to O are, respectively, 2.574(5), 2.634(5) and 2.517(5) Å. The inference of the short H-bond contacts is that the oxygen is strongly14,15
hydrogen-bonded to the three N atoms from the non-coordinating end of the host (Table 1). The length of the contact does not appear to depend on the secondary- or tertiary-nature of the N acceptor as the O⋯H⋯N(H) contact lies intermediate between the pair of O⋯H⋯N(Me) contacts. It must be noted that the Cu–O contact is also unusually short: at 1.860(5) Å, closer to what may be expected for OH− (or even O2−) than H3O+ coordination of Cu(II). In studies of anion coordination by these hosts, we have noted18 the facility with which water molecules may be polarised in the course of hydrogen bonding, particularly where multiply charged ions are involved. This process operates here, with the consequence that it is not possible to locate the origin of the third cationic charge with any certainty; indeed it appears likely that the charge is delocalised over the H-bonding array which comprises the tris-chelating N(H) system and the coordinated O. The apparent contradiction in the fact that the H-bonds, of average length 2.57 Å, are sufficiently short to justify assignment to charge-assisted O–H+⋯N hydrogen bonds deriving from water protonation while the unusually close Cu–O contact invites formulation as Cu–OH− or Cu–O2−, has to be resolved by considering that polarization has caused the water molecule to behave as though the O atom is negatively charged, while positive charge resides on or close to the N terminus of the hydrogen bond. The extreme limits, represented by Cu(II)L–H3O+, and Cu(II)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O with triprotonated ligand, have the virtue that trigonal symmetry, as required by the crystal structure, is preserved, but it seems clear that the actual situation lies somewhere between these extremes. The possibility that protonation has occurred elsewhere in the structure e.g., on the counterion or solvate water molecules, is less convincing as it does not serve to explain the unusual geometry in the neighborhood of the cation.
O with triprotonated ligand, have the virtue that trigonal symmetry, as required by the crystal structure, is preserved, but it seems clear that the actual situation lies somewhere between these extremes. The possibility that protonation has occurred elsewhere in the structure e.g., on the counterion or solvate water molecules, is less convincing as it does not serve to explain the unusual geometry in the neighborhood of the cation.
| Complex | Complex 2a | Complex 4 | Complex 5 |
|---|---|---|---|
| Formula | [Cu(L2)(H3O)](NO3)3·MeCN | [Cu(L3)(H3O)](ClO4)3 | [Zn(L1)(H3O)]BPh4(ClO4)2 |
| a Bridgehead N atom. b Minor component (33% occupancy). | |||
| M–O | 1.861(5) | 1.851(9) | 1.939(4) |
| 1.798(9)b | |||
| M–Nbra | 2.004(3) | 2.042(4) | 2.158(2) |
| 2.016(3)b | |||
| M–N | 2.169(3) | 2.322(3) | 2.277(2) |
| 2.252(3) | 2.278(2) | ||
| 2.387(3) | 2.321(2) | ||
| 2.247(3)b | |||
| 2.298(3)b | |||
| 2.444(3)b | |||
| O⋯H⋯N | 2.573(5) | 2.564(4) | 2.460(3) |
| 2.634(5) | 2.549(3) | ||
| 2.517(5) | 2.520(3) | ||
| 2.587(8)b | |||
| 2.536(7)b | |||
| 2.518(8)b | |||
| Nbr⋯Nbra | 6.818(4) | 6.837(7) | 7.035(3) |
The coordination of the Cu(II) cation is axially compressed trigonal bipyramidal, with irregular equatorial contacts to the secondary nitrogen, N3B 2.169(3), and pair of tertiary NMe donors: N3A and N3C at 2.387(3) and 2.252(3) respectively, averaging to 2.27 Å, as against axial contacts of 2.004(3) Å to Nbr and 1.860(5) Å to O1. The Nbr–Cu–O angle is just below linear at 176.04(13) and the N–Cu–O or N–Cu–Nbr angles deviate slightly from 90°, lying in the range 85–95°, in consequence of the siting of the Cu(II) cation slightly out of the N3 plane, as is normal2 for tren-derived cryptates. The Cu–N(Me) coordinate bonds are longer, by at least 0.15 Å, than those2,19 associated with sec-aminocryptates whether (mono- or di-) Cu(II) or average-valent dicopper. The extension in Cu–N bond length on methylation of the NH functions strongly suggests that weaker coordination, consequent on steric crowding at the tertiary amino site, is the price of alkylation.
Hexamethylated system
The hexamethylated cryptand, C24H54N8, L3, 3, can be obtained in a relatively pure state, by the reaction of excess of formaldehyde on the parent cryptand,11 L1, in formic acid solution. After reaction, it is contaminated only with the formaldehyde trimer, C3H6O3, from which it may be separated by fractional crystallization or by extraction with ethanol followed by size-exclusion chromatography. The infrared spectrum shows no absorption in the NH region, and elemental analysis together with mass spectral measurements confirm the nature and purity of the product as the neutral perN-methylated azacryptand. Lehn and coworkers,11 who originally synthesized the molecule using this route, studied its protonation behaviour potentiometrically. The protonation constants for the hexamethylated cryptand are similar to those reported8,11,20 for the unsubstituted parent cryptand; although the first pKa is noticeably smaller, and subsequent protonation constants marginally larger, under the same conditions, for the methylated versus unsubstituted cryptand. Although these effects are of marginal significance, they are consistent with established patterns in macrocyclic systems: the hexamethylated molecule behaves as a weaker base toward monoprotonation than the unsubstituted analogue, but for geometric reasons, perhaps linked to a cryptand expansion similar to that noted12 on methylation of analogous macrocyclic hosts, as a stronger base towards di- and subsequent protonation steps. Both methylated and unmethylated 2-carbon-linked cryptands are more basic toward monoprotonation than the 1,5-aromatic linked hosts we have studied previously21 but hexaprotonated level their basicity, as reflected by pK6, is much reduced due to a strong charge repulsion operating within the relatively small cavity. In these 2-C linked cryptands, the increase of acidity is more noticeable in the unsubstituted than in the perN-methylated cryptand, again suggesting a size expansion response to substitution, which attenuates the charge repulsion at higher protonation levels in 3.On treatment of the hexamethylated cryptand with copper(II) perchlorate in alcohol/acetonitrile solvent, followed by recrystallisation from DMF, insoluble blue–green hexagons of [CuL3(H3O)](ClO4)3·2H2O, 4, which show no NH absorption in the infrared spectrum, but a strong NMe feature, crystallise out slowly. Mass spectral analysis testifies to a monocopper formulation. Elemental analysis demonstrates the presence of three anions associated with the cation, but as expected, there was no intense electronic absorption as seen in average-valence dicopper. Structural characterisation of the perchlorate salt was sought in order to establish the nature of the complex.
This structure (Fig. 2) is very similar to, though somewhat more regular than, that discussed earlier (Fig. 1) for the tetramethyl analogue. The 50 : 50 disorder between the encapsulated copper and oxygen atoms is responsible for 32 symmetry in the cation (the disorder is not lost on reducing the symmetry). The half-occupancy copper atoms, accommodated in axially compressed trigonal bipyramidal geometry, occupy one of the N4-cap coordination sites, while an oxygen-centred species, (formally a protonated water molecule) occupies the other, making strong hydrogen bonds (2.564(4) Å) to the three amino functions which chelate it. The separation of the two guests at 1.851(9) Å, is of the same order as in the tetramethylated analogue (Fig. 1), certainly too short (despite the disorder) to be mistaken for a pair of copper ions in a copper–copper bond, and once again, difficult to reconcile with contact between a pair of positively charged (Cu2+ and H3O+) cations. It seems that, here again, polarisation of the OH bonds in the strongly H-bonding array has withdrawn positive charge from the oxygen atom causing it to behave, toward copper(II), like an anion. It is noticeable that the Cu(II)-tertN(Me) coordinate distance at 2.322(3) Å is much longer than is normal for Cu(II)–N contacts. Presumably this is a consequence of the increased rigidity of the guest molecule, now sterically unable to collapse in around the cationic guest to generate the more normal ≈ 2.1 Å Cu–N contacts. The pair of methylated hosts, L2 and L3 have very similar dimensions in their monoprotonated monocopper(II) complexes; the Nbr–Nbr distances lie at 6.817(4) and 6.837(4), respectively, (over 0.25 Å longer than this distance (6.52 Å) in the unsubstituted [Cu2L1]3+ cryptate), and the mutual separation of the N(R) atoms around the equatorial N3 plane averages to just over 4 Å in both cases. The conformations in the pair of complexes 2a and 4 are thus very similar. Fig. 3 illustrates the comparison with the conformation used in the average-valent dicopper cryptate of the unsubstituted host L1.
![Structure of the [CuL3(H3O)]3+ cation of 4. The hydrogen bonds are shown as solid lines and one component of the disorder has been omitted for clarity. Symmetry codes: A, −⅓ + y, ⅓ + x, ⅓
−
z; B, ⅔ + x
−
y, 1⅓ + x, ⅓
−
z; C, ⅔
−
x, ⅓
−
x + y, ⅓
−
z; D, 1 −
y, 1 + x
−
y, z; E, −x + y, 1 −
y, z.](/image/article/2006/DT/b602003h/b602003h-f2.gif) | ||
| Fig. 2 Structure of the [CuL3(H3O)]3+ cation of 4. The hydrogen bonds are shown as solid lines and one component of the disorder has been omitted for clarity. Symmetry codes: A, −⅓ + y, ⅓ + x, ⅓ − z; B, ⅔ + x − y, 1⅓ + x, ⅓ − z; C, ⅔ − x, ⅓ − x + y, ⅓ − z; D, 1 − y, 1 + x − y, z; E, −x + y, 1 − y, z. | ||
![Comparison of ligand conformations in [CuL3(H3O)]3+ (4, black bonds) and [Cu2L1]3+ (white bonds). The [CuL3(H3O)]3+ has been inverted for the fitting which was based on the Cu1 and the nitrogen atoms.](/image/article/2006/DT/b602003h/b602003h-f3.gif) | ||
| Fig. 3 Comparison of ligand conformations in [CuL3(H3O)]3+ (4, black bonds) and [Cu2L1]3+ (white bonds). The [CuL3(H3O)]3+ has been inverted for the fitting which was based on the Cu1 and the nitrogen atoms. | ||
There is no simple geometric explanation for the failure of L3 to encapsulate a pair of copper ions; the most obvious differences are found in the N–Cu distances and in the angles around the N–Cu coordination site, suggesting less effective coordination of the metal cation by the equatorial N donors. It is noticeable also that the methylated hosts utilize the more sterically favoured3 (owing to milder C–H repulsion) lel-conformation,22 rather than the ob-conformation22 adopted in the average-valence dicopper cryptate structure shown in Fig. 3.
We attempted incorporation of the metal cation into L3 under basic conditions, to see if the oxygen-centered species could be replaced by a second copper ion. When the metal coordination reaction is carried out under reflux with 1–2 stoichiometric equivalents of alcoholic KOH, or in the presence of two- to three-fold excess of NEt3, an increase in solubility of the product is noted, and the colour of the product is slightly darker and more bluish green. The crude product of this reaction analysed to a dicopper µ-hydroxocryptate of the ligand, which unfortunately we were unable to purify by crystallising from the reaction mixture. The most successful recrystallisation solvents have the effect of promoting the reprotonation equilibrium, so that the (more insoluble) protonated form is obtained to a greater or lesser degree on recrystallisation. The characteristic electronic23 and magnetic properties of average-valence dicopper failed to develop upon reaction with KOH: the only consequence of treatment with base apparently being formation of this µ-hydroxodicopper(II) centre. A colinear axial site24 for such an assembly seems unlikely given the steric constraints in this system; it seems more probable that the Cu2+ ions are accommodated in the cryptand faces as seen25 in the di- and tri-silver complexes of the unsubstituted ligand L1. Such coordination would generate bent ≈ 100–120° Cu–O–Cu angles in line with the moderate antiferromagnetic interaction evident in susceptibility measurements on this crude product. (see ESI† for temperature variation of susceptibility.) For mono µ-OH bridged dicopper complexes, a bridging angle of about 130° is known to generate moderate exchange (−2J) values26 in the region of 300 cm−1.
ESR and magnetic measurements were made to assist the characterization of the monocopper site in 4. ESR spectroscopy reveals a well-resolved spectrum characteristic of regular trigonal bipyramidal copper(II), as shown in Fig. 4, which demonstrates the existence of a dz2 ground state, with g⊥ > g∥ and A⊥ ≈ 1/3A∥. The ESR spectrum of 2a is basically similar to that of 4 but less regular (see ESI†). No ESR spectrum resembled the characteristic23 7-line pattern characteristic of axially symmetric average-valence dicopper.
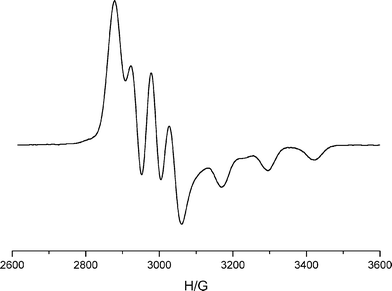 | ||
| Fig. 4 The X-band ESR spectrum of complex 4 as DMF glass at T = 170 K. Spectral parameters are g| = 1.995, A| = 128 G; g⊥ = 2.175, A⊥ = 51 G. | ||
Zinc complex of the unsubstituted cryptand ligand
The avidity of copper(II) for attachment to formally protonated water when encryptated in these small methylated azacryptands recalled a similar protonation behaviour encountered in the unsubstituted cryptand, L1, on encapsulation of zinc.4 No structures of the protonated cryptates of L1 were available at that time, but a suitable crystal of the tetraphenylborate/perchlorate salt, [ZnL1(H3O)](BPh4)(ClO4)2, 5, was later obtained, and the structure of the cation is now compared with those of 2a and 4. The zinc(II) ion, larger than copper(II) and lacking access to any lower oxidation state which would allow metal–metal bonding in an average-valent form, indeed encapsulates as a hydrogen bonded [M–O] species (Fig. 5). O⋯(H)⋯N hydrogen-bond distances in this complex, 5, are similar (Table 1) to those exhibited in the copper(II) N–Me cryptates, although the Zn–O and Zn–Nbr contacts are slightly longer in this less sterically constrained sec-aminocryptand host, where the overall Nbr–Nbr length of the cavity is 7.035(3) Å. The enhanced acidity of water coordinated to the Lewis-acidic cation, together with steric constraint enforcing close approach of these acidic water protons to the set of three amino-N atoms, again generates unusually strong H-bonding between the coordinated O atom and the three N(H) atoms (N⋯O average 2.51 Å). Such short contacts are known to develop when ionic character of the bond is high14,15,27 or where the pKa values of acid and base involved in the H-bonding interaction are well matched.17,28 Good matching of the pKa values is indeed likely in this case as the first pKa of the polyamino ligand L1 (at 10.70)20 is very similar29 to the pKa of the trenpodate of aquozinc(II), [Zn(tren)(H2O)]2+, 6, which represents the nearest available model for the “acid” function in this intramolecular H-bond. The pKa of 6 has been measured29 as 10.72. The fact the H-bonds, Table 1, are among the shortest so far recorded for O⋯H⋯N H-bonding14,15,17 also relies on the effect of the electrostatic interaction which develops, on charge transfer, between the three charged NH centres and the oxygen anion. At the shortest H-bond distances, polarization forces make a relatively important contribution to hydrogen bond energies, as do charge transfer terms.17 We believe that sterically enforced and charge-assisted hydrogen bonding is the origin of the strong O⋯H⋯N H-bond interaction seen in all three of the cryptates 2a, 4 and 5. The consequence of such strong H-bonding for activation of the zinc-hydroxide species active in catalysis of phosphate esters has been studied by Mareque-Rivas and coworkers who demonstrate, by potentiometric methods,30 the marked enhancement of acidity in aquoZn(II) podates of the tris 2-pyridylmethylamine series upon provision of appropriately positioned H-bond donors, and via kinetic measurements,31 an accompanying dramatic acceleration of phosphodiester cleavage catalysis in the same series.![Structure of the [ZnL1(H3O)]3+ cation of 5. The two-fold axis bisects the C3B–C3B′ and Zn–Zn′ vectors. The atoms of the second component of the disorder are shown dotted (50% occupancy of each site). Symmetry code, (′), 1 −
x, y,
−
z.](/image/article/2006/DT/b602003h/b602003h-f5.gif) | ||
Fig. 5 Structure of the [ZnL1(H3O)]3+ cation of 5. The two-fold axis bisects the C3B–C3B′ and Zn–Zn′ vectors. The atoms of the second component of the disorder are shown dotted (50% occupancy of each site). Symmetry code, (′), 1 −
x, y, ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) −
z.
−
z. | ||
The hydrogen bonding demonstrated in the structures of 2a, 4 and 5 is clearly much stronger than that associated with encapsulation of neutral water within the protonated but uncoordinated cryptand L1, as demonstrated by32 the structure of [H3L1(H2O)]3+·6H2O, 7 (Table 1). Here the average N⋯O contacts retaining the water guest within the cryptand host are much longer, at an average ≈ 2.85 Å. In the case of 7, where charge-assisted bonds from neutral water to the protonated nitrogens are present, we note that these are not particularly short, and are not in fact the shortest H-bonds in the cryptate, confirming the dominant role that steric enforcement of directionality together with cation-assisted polarization of water plays in generating the short H-bond contacts seen in 2a, 4 and 5. These short H-bonds are clearly responsible for considerable stabilisation of the [M–O] assembly. In contrast, in the structure of the aquozinc(II)tren-podate,29 the Zn–Ow contact is of normal length at an average of 2.121(4) Å while the Zn–N coordination distances are shorter than those in 5 by > 0.20 Å. A recent report of a Cu(II) aminopodate with neutral water axial33 shows the Cu–Ow contact is significantly longer than in the cryptates 2a and 4, at 1.964(4) Å, and here too, the M–N coordination distances fall in the much shorter and more usual range of 2.08–2.17 Å. It appears, then, that steric constraints in the methylated hosts serve to destabilize aminocryptand coordination of metal cations at the same time as stabilizing the encapsulation of protonated water.
In the case of the Cu(II) N–Me cryptates studied here, the combination of stabilization provided by hydrogen bonding and destabilization of second-cation coordination is sufficient to compete successfully with generation of any potential one-electron copper–copper bond. We have recently noted that the relative stability of mononuclear versus dinuclear average-valent copper within L1 is finely balanced and solvent dependent.19 The solvation energy of acetonitrile/copper(I) is sufficient to set up a competition which is almost isoenthalpic, the temperature dependence of the stability constant being governed by entropic terms. In the present system, even where deprotonation is favoured by increasing the basicity of the medium, no tendency to adopt the average-valent dicopper structure in solution has been demonstrated.
Conclusion
The steric constraints imposed by these small dinucleating cryptands once again serve to enforce an unusual bonding situation upon a pair of encapsulated guests. The accommodation of a coordinated water molecule within the restricted cavity locates it so close to the dipositively charged Lewis-acidic cation as to result in severe polarisation of the OH bonds, generating a bonding situation closer to [M–O2−] than to [M–H3O+]. The effective negative charge at the oxygen atom together with the proximity and appropriate geometry of the set of three partially charged NH atoms results in three very efficient ± charge-assisted H-bonding interactions, correlated through their connection to the common O atom.These cryptand hosts are thus naturally adapted to recognize and stabilize a [M–O] assembly. This could have implications for the generation of biologically important terminal oxoligated higher-valent iron and manganese species.34,35 Although our initial concern in the present context was that the stability of the [M–O] assembly within these hosts made it impossible to replace the [Cu–O] guest by the expected copper–copper bonded assembly, the encapsulated [M–O] assembly now appears as an important priority in its own right, particularly regarding any potential for catalysis of hydrolysis reactions. The important differences from the tripodal systems currently under study,30,31 in respect of H-bond strength, access to the active site and lability of the coordinated “hydroxo” species, promise to generate new and valuable information on the mechanism of these reactions.
Experimental
Synthesis
The ligand L1 was synthesized as described earlier.3,19It appears difficult to deprotonate the ligand salt formed in this way. Used in situ with protonated ligand and copper(II) salts, neither acetate nor triethylamine succeeds in formation of neutral ligand and the use of alcoholic KOH generates ill-defined insoluble copper(II) hydroxo species.
[Cu2HI2L2(H3O)](NO3)3·4H2O, 2 and [CuL2(H3O)](NO3)3·4H2O·MeCN, 2a
0.33 g (0.5 mM) of 1 was dissolved in 10 cm3 ethanol. To this was added 0.28 g (1.46 mM) of copper nitrate in 10 cm3acetonitrile. After 2 h reflux, the solution was evaporated to dryness. A dark green–brown hygroscopic powder was obtained, which analysed to Cu2HL2I2(NO3)3·4H2O, but was not investigated further, because of its instability. The mode of coordination and siting of the Cu(II) cations, i.e. whether intra- or extra-cavity has thus not been established. %CHN (calculated values in parenthesis) 24.49 (24.81); 4.90 (5.48); 14.90 (14.47). Attempts at purification of this sample, by recrystallisation from DMF, resulted in an immediate colour change to deep bottle green. Suitable crystals of 2a for X-ray characterization were obtained by slow diffusion of THF into the DMF solution.Selected IR absorptions: 3436 br s; 3174, 3073, w; 2869, m; 1636 mw, 1383 vs. %CHN (calculated values in parenthesis) 34.87 (34.44); 7.45 (7.95); 20.45 (20.09) ESMS (rel. intensities in parenthesis) m/z 551 (68) CuL2NO3+; 570 (6) CuL2NO3(H2O)+; 636(7) CuL2(NO3)2(H3O)+
The product of treatment of 1 with copper(II) perchlorate (which, although isolable in an apparently crystalline form, fails to diffract X-rays) is likewise a complex of the protonated ligand. Recrystallisation from THF/DMF yields blue aqua spars of [CuL2(H3O)](ClO4)3·2H2O, 2b, analogous to 2a %CHN (calculated values in parenthesis) 31.61 (31.35); 6.29 (6.58); 12.74 (13.29). Selected IR absorptions: 3431 br s; 3245 w; 2874 m; 1653 m, 1476 m, 1091 vs, 625 m.
%CHN (calculated values in parenthesis) 58.52 (58.29); 9.68 (9.72); 22.74 (22.67) ESMS (relative intensities in parenthesis) mass 455 appears as a monopositively charged cluster at ≈ 455 (44%) and dipositively charged at ≈ 228 (100%). 1H NMR spectrum was consistent with that reported in reference 11.
A cluster at m/z 797 in the ESMS of unrecrystallised 4 indicates the presence of this dicopper µ-hydroxo product in the bulk sample of 4.
Magnetic susceptibility determination (µ(80) = 1.14 µB; µ(273) = 1.45 µB) of this product demonstrates the presence of a moderately weak antiferromagnetic interaction. However, the data do not fit the Bleaney–Bowers equation (see ESI†). The ESR spectrum shows a mononuclear spectrum similar to that of 4, demonstrating that hydrolysis has taken place upon dissolution in DMF.
Cryptate 5, [ZnL1(H3O)](BPh4)(ClO4)2 was made in the form of the triperchlorate salt as described earlier4 and converted to the BPh4 salt by redissolution in MeCN, followed by NaBPh4 addition as follows: to 0.005 mmole [ZnL1(H3O)](ClO4)3 in 3 cm3 MeCN is added 0.011 mmole NaBPh4 in 3 cm3 EtOH. X-Ray quality crystals are obtained on slow evaporation in air.
Magnetism and ESR spectroscopy
Magnetic susceptibility measurements were made using an Oxford Instruments Faraday Magnetometer with variable temperature facility. Diamagnetic corrections determined from Pascal constants were applied. ESR spectra were recorded with a Varian E109 X-band spectrometer equipped with a liquid nitrogen-cooled cryostat.X-Ray crystallography
Data for complexes 2a and 4 were collected at 150(2) K on a Bruker SMART 1000 CCD diffractometer, while data for complex 5 were collected on station 9.8 of the SRS, Daresbury. All three structures were solved by direct methods and refined on F2 using all the reflections.36 All the non-hydrogen atoms were refined using anisotropic atomic displacement parameters and hydrogen atoms bonded to carbon were inserted at calculated positions using a riding model. In each case, disorder of the metal and oxygen atoms prevented location of the protons involved in the hydrogen bonding inside the crypt and these were not included in the model. In addition to the disorder in the cation described in the Discussion, each structure showed some disorder among the counter ions which was modeled conventionally. Parameters for data collection and refinement are summarised in Table 2.CCDC reference numbers 298081–98083.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b602003h.
| Complex | Complex 2a | Complex 4 | Complex 5 |
|---|---|---|---|
| Formula | [Cu(L2)(H3O)](NO3)3·MeCN | [Cu(L3)(H3O)](ClO4)3 | [Zn(L1)(H3O)]BPh4(ClO4)2 |
| Empirical formula | C24H53CuN12O10 | C24H57Cl3CuN8O13 | C42H65BCl2N8O9Zn |
| Formula weight | 733.32 | 835.67 | 973.10 |
| Temperature/K | 150(2) | 150(2) | 150(2) |
| Wavelength/Å | 0.71073 | 0.71073 | 0.69230 |
| Crystal system | Monoclinic | Rhombohedral | Monoclinic |
| Space group | P2(1)/n | R32 | C2/c |
| a/Å | 11.2153(7) | 10.1113(12) | 13.5719(5) |
| b/Å | 14.4404(8) | 10.1113(12) | 32.9185(7) |
| c/Å | 21.0213(12) | 29.655(5) | 10.4070(4) |
| α/° | 90 | 90 | 90 |
| β/° | 99.298(1) | 90 | 96.140(2) |
| γ/° | 90 | 120 | 90 |
| Volume/Å3 | 3359.7(3) | 2625.7(6) | 4622.8(3) |
| Z | 4 | 3 | 4 |
| Absorption coeff./mm−1 | 0.720 | 0.926 | 0.709 |
| Refl. collected | 23760 | 6242 | 21556 |
| Ind. refl. [Rint] | 5926 [0.0229] | 1042 [0.0351] | 5713 [0.0471] |
| R1, wR2 [I > 2σ(I)] | 0.0581, 0.1380 | 0.0367, 0.0957 | 0.0471, 0.1125 |
| R1, wR2 (all data) | 0.0661, 0.1419 | 0.0407, 0.0977 | 0.0615, 0.1164 |
Acknowledgements
We thank BBSRC for funding (DF) and EPSRC for access to the Mass Spectrometric service at Swansea, we also thank CCRC for use of facilities at station 9.8 at Daresbury. JN thanks the Leverhulme Trust for an Emeritus Fellowship which enabled the completion of this work and its preparation for publication. Loughborough University is thanked for the award of an Honorary Professorial Fellowship to JN.References
- A. M. Sargeson, Pure Appl. Chem., 1978, 50, 905 CrossRef CAS.
- J. Nelson, V. McKee and G. G. Morgan, Prog. Inorg. Chem., 1998, 47, 162.
- J. A. Thompson, M. E. Barr, D. K. Ford, L. A. Silks, B. J. McCormick and P. H. Smith, Inorg Chem., 1996, 35, 2025 CrossRef CAS; P. H. Smith, M. E. Barr, J. R. Brainard, D. K. Ford, H. Freiser, S. Muralidharar, S. D. Reilly, R. R. Ryan, L. A. Selles and W. H. Yu, J. Org. Chem., 1993, 58, 7939 CrossRef CAS.
- J. L. Coyle, M. G. B. Drew, C. J. Harding, J. Nelson and R. M. Town, J. Chem. Soc., Dalton Trans., 1997, 1123 RSC.
- N. Martin, V. McKee and J. Nelson, Inorg. Chim. Acta, 1994, 218, 5 CrossRef CAS.
- M. A. Hossain, J. M. Llinares, N. W. Alcock, D. Powell and K. Bowman-James, J. Supramol. Chem., 2002, 2, 143 Search PubMed.
- B. Dietrich, J. M. Lehn, J. Guilhem and C. Pascard, Tetrahedron Lett., 1989, 30, 4128.
- S. D. Reilly, G. R. K. Khalsa, D. K. Ford, J. R. Brainard, B. P. Hay and P. H. Smith, Inorg. Chem., 1995, 34, 569 CrossRef CAS.
- S. Iwata, C. Ostermeier, B. Ludwig and H. Michel, Nature, 1995, 376, 660 CrossRef CAS; T. Tsikihara, H. Aoyama, E. Yamashita, T. Tomazaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono and S. Yoshikawa, Science, 1995, 269, 11069.
- K. Brown, M. Tegoni, M. Prudencio, A. S. Periera, S. Besson, J. J. Moura, I. Moura and C. Cambillau, Nature Struct. Biol., 2000, 7, 191 CrossRef CAS.
- B. Dietrich, B. Dilworth, J. M. Lehn, J. P. Souchez, M. Cesario, J. Guilhem and C. Pascard, Helv. Chim. Acta, 1996, 79, 569 CrossRef CAS.
- A. Andres, C. Bazzicalupi, A. Bencini, A. Bianchi, V. Gusi, E. Garcia-Espana, C. Giorgi, N. Nardi, P. Paoletti, J. A. Ramirez and B. Valtancoli, J. Chem. Soc., Perkin Trans. 2, 1994, 2367 RSC.
- P. W. Bernhardt, J. M. Harrowfield, D. C. R. Hockless and A. M. Sargeson, Inorg. Chem., 1994, 33, 5659 CrossRef CAS.
- G. Gilli and P. Gilli, J. Mol. Struct., 2000, 552, 1 CrossRef CAS.
- In the unified hydrogen bond theory of Gilli and Gilli,14 an N⋯O contact of 2.532 Å is reported as the minimal distance for a N–H⋯O− charge-assisted hydrogen, described in the biochemical literature16 as a salt bridge. In uncharged systems,17 the range of NH⋯O contacts is listed as 2.70–3.09 Å, while the range of OH⋯N contacts is 2.84–3.03 Å.
- G. A. Jeffrey, Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- T. Steiner, Angew. Chem., 2002, 41, 48 CAS.
- J. Nelson, V. McKee and R. M. Town, Oxoanion Selectivity with Protonated Azacryptate Hosts: the Influence of Hydration on Structure and Stability, in Macrocyclic Chemistry, ed. K Gloe, Springer, Dordrecht, 2005, pp. 189–203 Search PubMed.
- J. Coyle, A. J. Downard, C. J. Harding, R. Herbst-Irmer, V. McKee and J. Nelson, Dalton Trans., 2004, 2357 RSC.
- pKa values for azacryptands taken from ref. 11; (0.1 M (Me4N)OTs, 25° C): for L1: pK1 10.70; pK2, 9.20; pK3, 7.50; pK4 5.50; [pK5 + pK6] 5.10; and for L3: pK1 10.05; pK2 9.70; pK3 8.20; pK4 5.75; pK5 3.40; pK6 2.30.
- F. Arnaud-Neu, S. Fuangswasdi, B. Maubert, V. McKee, J. Nelson and M.-J. Schwing, Inorg. Chem., 2000, 39, 573 CrossRef CAS.
- As defined in reference 2, p. 210.
- C. J. Harding, J. Nelson, M. C. R. Symons and J. Wyatt, J. Chem. Soc., Chem. Commun., 1994, 2499 RSC.
- C. J. Harding, V. McKee, J. Nelson and L. Qin, J. Chem. Soc., Chem. Commun., 1993, 1768 RSC.
- V. McKee, J. Nelson, D. J. Speed and R. M. Town, J. Chem. Soc., Dalton Trans., 2001, 3641 RSC.
- K. Patra, M. Ray and R. Mukherjee, Polyhedron, 2000, 19, 1423 CrossRef CAS.
- T. Steiner, C. C. Wilson and I. Majerz, Chem. Commun., 2000, 1231 RSC.
- P. Gilli, V. Bertolasi, L. Pretto, A. Lycka and G. Gilli, J. Am. Chem. Soc., 2002, 124, 13554 CrossRef CAS.
- M. Ibrahim, N. Shimomura, K. Ichikawa and M. Shiro, Inorg. Chim. Acta, 2001, 313, 125 CrossRef CAS.
- J. C. Mareque-Rivas, R. Prabaharan and R. T. M. de Rosales, Chem. Commun., 2004, 76 RSC.
- G. Feng, J. C. Mareque-Rivas, R. T. M. de Rosales and N. H. Williams, J. Am. Chem. Soc., 2005, 127, 13470 CrossRef CAS.
- D. T. Farrell, J. Nelson and V. McKee, Acta Crystallogr., Sect. E, 2005, E61, 081.
- M. Schatz, M. Becker, O. Walter, G. Liehr and S. Schindler, Inorg. Chim. Acta, 2001, 324, 173 CrossRef CAS.
- C. J. Elsevier, J. Reedijk, P. H. Walton and M. D. Ward, Dalton Trans., 2003, 10, 1869 RSC.
- C. E. MacBeth, B. S. Hammes, V. G. Young, Jr. and A. S. Borovik, Inorg. Chem., 2001, 40, 4733 CrossRef CAS.
- G. M. Sheldrick, SHELXTL Version 6.12, Bruker AXS, Madison WI, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Magnetic and ESR data. See DOI: 10.1039/b602003h |
| This journal is © The Royal Society of Chemistry 2006 |