The enzymatic activation of coenzyme B12
Kenneth L. Brown*
Department of Chemistry and Biochemistry, Ohio University, Athens, OH 45701, USA. E-mail: brownk3@ohiou.edu; Fax: +1 740-593-0148; Tel: +1 740-593-1737
First published on 7th February 2006
Abstract
The enzymatic “activation” of coenzyme B12 (5′-deoxyadenosylcobalamin, AdoCbl), in which homolysis of the carbon–cobalt bond of the coenzyme is catalyzed by some 109- to 1014-fold, remains one of the outstanding problems in bioinorganic chemistry. Mechanisms which feature the enzymatic manipulation of the axial Co–N bond length have been investigated by theoretical and experimental methods. Classical mechanochemical triggering, in which steric compression of the long axial Co–N bond leads to increased upward folding of the corrin ring and stretching of the Co–C bond is found to be feasible by molecular modeling, but the strain induced in the Co–C bond seems to be too small to account for the observed catalytic power. The modeling study shows that the effect is a steric one which depends on the size of the axial nucleotide base, as substitution of imidazole (Im) for the normal 5,6-dimethylbenzimidazole (Bzm) axial base decreases the Co–C bond labilization considerably. An experimental test was thus devised using the coenzyme analog with Im in place of Bzm (Ado(Im)Cbl). Studies of the enzymatic activation of this analog by the B12-dependent ribonucleoside triphosphate reductase from Lactobacillus leichmannii coupled with studies of the non-enzymatic homolytic lability of the Co–C bond of Ado(Im)Cbl show that the enzyme is only slightly less efficient (3.8-fold, 0.8 kcal mol−1) at activating Ado(Im)Cbl than at activating AdoCbl itself. This suggests, in agreement with the modeling study, that mechanochemical triggering can make only a small contribution to the enzymatic activation of AdoCbl. Another possibility, electronic stabilization of the CoII homolysis product by compression of the axial Co–N bond, requires that enzymatic activation be sensitive to the basicity of the axial nucleotide. Preliminary studies of the enzymatic activation of a coenzyme analog with a 5-fluoroimidazole axial nucleotide suggest that the catalysis of Co–C bond homolysis may indeed be significantly slowed by the decrease in basicity.
 Kenneth L. Brown Kenneth L. Brown | Ken Brown was born in Philadelphia, PA. He earned a BS in Biochemistry from the University of Chicago in 1968 and a PhD in Biochemistry from the University of Pennsylvania in 1971. After working as a NIH Postdoctoral Fellow in the Department of Biochemistry and Biophysics at the University of California at Davis in Lloyd Ingraham's laboratory, he joined the faculty in the Department of Chemistry at the University of Texas, Arlington, in 1975. In 1990 he moved to Mississippi State University as Professor and Head of the Department of Chemistry, and in 1996 he moved to Ohio University where he is now Professor and Chair of the Department of Chemistry and Biochemistry. |
Introduction
The coenzyme form of vitamin B12 (5′-deoxyadenosylcobalamin, AdoCbl, Fig. 1) is known to be involved in the catalysis of about a dozen enzymatic reactions involving 1,2-intramolecular substrate rearrangements or the reduction of ribonucleotides to deoxyribonucleotides. The initial event in the catalytic cycle of all AdoCbl-dependent enzymes (the so-called “activation” of AdoCbl) is the enzyme-induced homolysis of the carbon–cobalt bond of the coenzyme to form the CoII species (cob(II)alamin) and a radical at the active site, a reaction which can be catalyzed by these enzymes by 109- to 1014-fold.1,2,3 Despite 50 years of active research on AdoCbl, the mechanism of enzymatic activation of AdoCbl is not yet known for any AdoCbl-dependent enzyme and remains one of the outstanding problems in bioinorganic chemistry.4,5 | ||
| Fig. 1 Structure and numbering scheme for AdoCbl and the structure of analogs with altered axial nucleotides. | ||
These enzymes are now known to fall into three classes, distinguished by the nature of the migrating group and the nature of the substituent on the carbon atom to which this group migrates.6 The class I enzymes are mutases (methylmalonylCoA mutase,7 glutamate mutase,8 2-methyleneglutarate mutase,9 and isobutyrylCoA mutase10) which catalyze carbon skeleton rearrangements. The class II enzymes include the eliminases (diol dehydratase,11 glycerol dehydratase,12 and ethanolamine ammonia lyase13) and the AdoCbl-dependent ribonucleotide reductases.14 The less well studied class III enzymes are aminomutases (D-lysine-5,6-aminomutase and D-ornithine mutase)15 which catalyze amino group migrations and require pyridoxal phosphate in addition to AdoCbl. Important differences between the class I and class II enzymes include the character of the biradical EPR spectra obtained by cryotrapping the enzymes during turnover, the binding mode of the coenzyme, and the specificity for the coenzyme.
For the class I enzymes, the EPR spectrum of the biradical intermediate shows hyperfine coupling and a zero-point crossing g value between the CoII and organic radical g values.16 These spectra result from relatively strong spin–spin coupling between the cobalt unpaired spin and that of the organic radical. In contrast, the class II enzymes exhibit weak coupling so that the CoII and organic radical features are resolved. The CoII signal is generally broad and featureless at g ∼ 2.2, and a sharper organic radical feature appears near g = 2.
The class I AdoCbl-dependent enzymes are known from EPR studies17,18 and X-ray crystallography19,20 to bind AdoCbl in a base-off conformation, with the α (or lower) axial ligand position occupied by the imidazole moiety of a histidine residue and the pendent 5,6-dimethylbenzimidazole nucleotide (Bzm) buried in a hydrophobic pocket. In contrast, EPR studies21,22,23 and X-ray crystallography24,25 have conclusively demonstrated that the class II enzymes bind AdoCbl in the base-on conformation, with the Bzm nucleotide coordinated to the cobalt atom. Both of the class III aminomutases are now known to bind AdoCbl in the base-off mode, similar to the class I mutases.26,27
The other important characteristic distinguishing the class I and class II enzymes is their specificity for AdoCbl. The class II enzymes, including ribonucleotide reductase,28 diol dehydratase,29 and glycerol dehydratase,30 are remarkably promiscuous in tolerating structural alterations in the coenzyme. A large number of AdoCbl analogs with structural alterations in the adenine and ribose moieties, the corrin, and its side chains, and in the axial nucleotide are partially active coenzymes with these enzymes (although other AdoCbl analogs are inactive, and act as inhibitors). In stark contrast, the class I glutamate mutase is nearly absolutely specific for AdoCbl itself with only one of 23 structural analogs (many of which are partially active coenzymes with class II enzymes) investigated showing any activity at all,31 and MMCoA mutase can utilize a single coenzyme analog with an altered axial nucleotide structure,32 but cannot function with analogs altered in the adenosyl moiety.33 These observations suggest that the mechanism of activation may not be the same for all AdoCbl-dependent enzymes, a surprising possibility that was not even dreamed of just a few years ago.
Coenzyme activation is a simple bond dissociation reaction
With the possible exception of the B12-dependent ribonucleotide reductase (vide infra) and the class I mutases, in which Co–C bond cleavage may be concerted with a subsequent hydrogen atom transfer, the enzymatic activation of AdoCbl by Co–C bond homolysis is a simple bond dissociation reaction. Although non-enzymatic, thermal homolysis of AdoCbl and other alkylcobalt corrinoids leads to a solvent caged radical pair, the diffusive separation of which is the rate determining step,34,35,36 this cannot be the case at an enzyme active site, where there is little or no solvent, and diffusive separation of a radical pair is unlikely. Since there is no compensatory bond formation, the enzymatic activation of AdoCbl is a simple bond dissociation process, the “free energy of activation” is the bond dissociation energy (30 kcal mol−1),34 and the reaction coordinate diagram is simply the portion of the Morse potential curve that raises with increasing distance (Fig. 2). As this reaction coordinate diagram has no maximum, there is no transition state, and the reaction can only be catalyzed by destabilizing the reactant, stabilizing the products, or a combination of the two. | ||
| Fig. 2 (A) Hypothetical Morse potential curve for the Co–C bond of AdoCbl, showing the measured bond dissociation energy. (B) Hypothetical reaction coordinate diagram for the enzyme-induced Co–C bond homolysis of AdoCbl showing the necessary stabilization of the products or destabilization of the reactant needed to explain the catalysis. | ||
Ribonucleoside triphosphate reductase from L. leichmannii
Due to its physical and catalytic properties, the AdoCbl-dependent ribonucleoside triphosphate reductase (RTPR) from Lactobacillus leichmannii is an ideal enzyme for studies of AdoCbl activation. Unlike the other well characterized AdoCbl-dependent enzymes, most of which are multimers of dissimilar subunits of native molecular weight over 100 kDa, the leichmannii RTPR is a relatively small, supposedly monomeric allosteric enzyme of 82 kDa37 with a diffusion coefficient of 6.4 × 10−7 cm2 s−1.38 We can thus estimate that its hydrated radius is about 38 Å and its rotational correlation time is about 50 ns.The enzyme's catalytic properties are also important. While a low molecular weight thioredoxin is the natural reductant, dithiols such as dihydrolipoate are also catalytically active electron donors.38 In addition, the enzyme catalyzes the exchange of hydrogens between the 5′-methylene of AdoCbl and solvent,39 and has been shown to catalyze kinetically competent activation of AdoCbl to cob(II)alamin by EPR40,41 and stopped-flow measurements.41,42 Unlike all other AdoCbl-dependent enzymes, the bisubstrate RTPR catalyzes both the exchange reaction and AdoCbl activation in an incomplete system consisting of enzyme, dithiol reductant, AdoCbl, and the allosteric effector dGTP, allowing activation to be observed in the absence of turnover. Moreover, rapid quench EPR40,41 and stopped flow UV-visible41,42 experiments show that enzymatically-induced generation of the cob(II)alamin UV-visible and EPR spectra occur with the same rate constant (ca. 40 s−1 at 37 °C). Kinetic studies of the catalysis of AdoCbl homolysis in the absence of a reducible NTP substrate2,43 coupled with detailed studies of the non-enzymatic thermal homolysis of AdoCbl3 show that the enzyme catalyzes Co–C bond homolysis by a factor of 1.6 × 109 (13 kcal mol−1) at 37 °C, and shifts the equilibrium towards homolysis products by an astonishing 2.9 × 1012 (17.7 kcal mol−1) as a result of differential binding of AdoCbl and the homolysis products.3
The EPR spectrum observed after rapid freeze quenching of reaction mixtures containing RTPR, AdoCbl, a dithiol, and the allosteric effector dGTP is a complex pattern centered at g = 2.119 that has been interpreted as being due to weak coupling between cob(II)alamin and another radical, once thought to be the Ado˙ radical.42 Elegant experiments from Stubbe's lab41 have shown that this characteristic EPR spectrum is due to the presence of cob(II)alamin and a thiyl radical derived from the essential44 active site residue, Cys408. Thus, incorporation of [β-2H2]-cysteine into cloned37 RTPR produces significant sharpening of the EPR spectrum, demonstrating the presence of a cysteine-based thiyl radical. Hence, the products of AdoCbl activation actually observed at the active site are cob(II)alamin and a thiyl radical, although it is not known if the Ado˙ radical is formed first and abstracts a hydrogen from Cys408, or if carbon–cobalt bond homolysis and hydrogen atom transfer are concerted. An important paper by Frey, Stubbe and coworkers45 has shown that Co–C bond cleavage can occur without H-atom transfer. However, this work does not settle the question of whether Co–C bond cleavage and H-atom transfer are concerted or stepwise.
Mechanisms of activation of AdoCbl
As described above, for those enzymes in which AdoCbl activation is a simple bond dissociation reaction, there is no definable transition state, and catalysis can only be obtained by destabilizing the reactant, stabilizing the products of Co–C bond homolysis, or a combination of these two effects. For RTPR, an additional mechanism is possible if Co–C bond cleavage and H-atom transfer from the essential active site Cys408 residue to the emerging Ado˙ radical is concerted. In this case, there is a transition state which is stabilized by the partial H-atom transfer, since the thiyl radical that is generated is more stable than the Ado˙ radical (Scheme 1). However, the energetics of the radicals involved suggests that such transition state stabilization would not be very large. | ||
| Scheme 1 | ||
It has long been thought that the flexibility of the corrin ring is important and may be exploited by these enzymes to labilize the Co–C bond by increasing the “upward” fold of the corrin to sterically accelerate Co–C bond cleavage.46,47,48,49,50,51,52,53 One such mechanistic proposal, the mechanochemical triggering hypothesis, envisions enzymatic compression of the long (2.24 Å)54 axial Co–N bond and a consequent increase in the steric interactions of the bulky Bzm axial nucleotide with the underside of the corrin as the means by which an enzyme could engender an increase in the upward fold of the corrin ring.50,52,53,55 Mechanisms prominently involving the axial Bzm nucleotide have long been of interest since they would provide a rationale for cobalamin being the only naturally occurring pentadentate metallocycle. Whether such a mechanism could be used by class I enzymes, which displace the Bzm ligand of AdoCbl with a less bulky histidine residue, remains unclear.
Another possibility is that manipulation of the axial Co–N bond by the enzyme leads to stabilization of the cob(II)alamin product state.56 Although few direct comparisons are available, the axial Co–N bonds of cobalamins may be abnormally long. For example, the axial Co–N bond in CH3Cbl57 is 0.1 Å longer than that in methyl(1,5,6-trimethylbenzimidazole)cobaloxime.58 If this is the result of steric interactions between the bulky Bzm ligand and the corrin, it implies that the axial bond orbital overlap is non-ideal. Thus, enzyme-induced steric compression of the axial Co–N bond in cob(II)alamin could provide electronic stabilization of this homolysis product.
Modeling mechanochemical triggering
The feasibility of mechanochemical triggering has been studied by molecular mechanics,59,60 an excellent example of the utility of such methods. This was accomplished by varying the value of the strain-free bond length, lo, for the axial Co–N bond to provide models with various axial Co–N bond lengths, an experimentally impossible feat. The results are shown in Fig. 3 as plots of the corrin ring fold angle (Fig 3A), the Co–C bond length (Fig. 3B), and the Co–C–C bond angle (Fig. 3C) vs. the axial Co–N bond length for AdoCbl and the analog in which the axial Bzm ligand is replaced by imidazole (Ado(Im)Cbl). As shown on the left panel of the figure, the model of AdoCbl accurately duplicates the essential features of the coenzyme when the axial Co–N bond length of the model (2.231 Å) is essentially the same as that found in the neutron diffraction structure of AdoCbl (2.24 Å).54 Thus, at this axial Co–N distance, the model has a corrin ring fold angle of 14.9°, a Co–C bond distance of 2.041 Å and a Co–C–C bond angle of 122.6°, while the neutron diffraction structure gives 14.6°, 2.04 Å, and 123°, respectively.54 | ||
| Fig. 3 Dependence of (A) the corrin ring fold angle, (B) the Co–C bond length, and (C) the Co–C–C bond angle on the axial Co–N bond length in molecular models of AdoCbl (●) and Ado(Im)Cbl (■). The panel on the right shows cartoons depicting the motions investigated, and the panel on the left compares the model results for AdoCbl to the neutron diffraction structure of AdoCbl54 when the axial Co–N bond length of the model is the same as that of the diffraction structure. | ||
Each of the three modeled parameters is independent of the axial Co–N bond length when this bond length is greater than ca. 3 Å. However, as the axial Co–N bond length is compressed below 3 Å, the fold angle, the Co–C bond length, and the Co–C–C bond angle all increase, indicating progressive upward folding of the corrin ring, and weakening of the Co–C bond. Importantly, although the same pattern is followed in the Ado(Im)Cbl analog, the increase in all three parameters is always smaller than that for AdoCbl itself, demonstrating the importance of the steric bulk of the axial ligand to the mechanochemical effect.
These models clearly confirm the feasibility of classical mechanochemical triggering and demonstrate that it is a steric effect of the bulk of the axial ligand. Relative total strain energies of the models permit estimation that compression of the axial Co–N bond of AdoCbl from its normal value of 2.23 Å54 to 1.75 Å (less than 0.2 Å shorter than that in H2OCbl+)61 requires 19 kcal mol−1 of energy, which clearly could be supplied by binding energy, as proteins are known to be capable of binding Cbl's extremely tightly (e.g., Kb = 5.0 × 1016 M−1, 23.7 kcal mol−1 at 37 °C, for CNCbl binding to the B12-binding protein, haptocorrin62). However, at this axial Co–N bond length, the amount of Co–C bond distortion is small (a 0.035 Å increase in Co–C bond length and a 1.4° increase in Co–C bond angle), and previous work63 suggests that this distortion would contribute only a few kcal mol−1 reduction in the free energy of activation, far short of the 13 kcal mol−1 observed for RTPR.2,3
Steady state enzymatic activity of Ado(Im)Cbl and Ado(Bzim)Cbl
The fact that molecular modeling shows that mechanochemical triggering depends on the steric bulk of the axial nucleotide is quite important for it allows an experimental test of this mechanism using coenzyme analogs with altered axial nucleotides. Ado(Im)Cbl and the coenzyme analog with benzimidazole in place of the normal 5,6-dimethylbenzimidazole axial base (Ado(Bzim)Cbl) have been synthesized by adenosylation of the cyano derivatives obtained from “guided biosynthesis”64,65,66,67 using fermentation of Propionibacterium shermanii on media supplemented with imidazole or benzimidazole, respectively, employing a modification of the method of Renz.66,67 Steady state kinetics with RTPR showed that both coenzyme analogs supported catalysis with the same Vmax as AdoCbl.64 However, while the apparent Km for Ado(Bzim)Cbl (0.14 ± 0.01 µM)68 was essentially the same as that for AdoCbl (0.18 ± 0.01 mM),2 the apparent Km for Ado(Im)Cbl (13 ± 1 µM) was some 70-fold higher.68Enzymatic activation of Ado(Im)Cbl and Ado(Bzim)Cbl
The kinetics of formation of the cob(II)alamin analog at 37 °C upon mixing RTPR and Ado(Im)Cbl or Ado(Bzim)Cbl were measured68 spectrophotometrically with a stopped flow apparatus using dithiothreitol (DTT) as the reductant, dGTP as the allosteric effector, and no NTP substrate41,42 as previously determined for AdoCbl itself.2 The results (Fig. 4, for Ado(Bzim)Cbl) were qualitatively similar to those previously obtained for AdoCbl.2 As before, the observed first-order rate constants for the rapid, reversible formation of cob(II)alamin were independent of the enzyme concentration, but the absorbance change, ΔA, indicative of the extent of formation of the cob(II)alamin species, increased with RTPR concentration and appears to “saturate.” The simplest interpretation of these results is that rapid coenzyme binding (Kb) is followed by the observable, reversible cleavage of the carbon–cobalt bond (kf and kr, eqn (1)). | (1) |
 | ||
| Fig. 4 (A) Dependence of the absorbance change, ΔA, accompanying the reversible formation of (Bzim)cob(II)alamin from Ado(Bzim)Cbl on the initial concentration of RTPR at 37 °C. The solid line is a least squares fit to a binding isotherm, from which the values ΔA∞ = 0.126 ± 0.004 and Kb = 1.72 ± 0.20 × 104 M−1 were obtained. (B) Dependence of the observed rate constant for enzymatic formation of (Bzim)cob(II)alamin from Ado(Bzim)Cbl on the initial concentration of RTPR at 37 °C. The solid line is the average value (24.2 ± 1.4 s−1) and the dashed lines are plus or minus one standard deviation. | ||
As previously,2 the dependence of ΔA on enzyme concentration allows calculation of the binding constant, Kb, for the Ado(L)Cbl coenzyme to the enzyme, and the maximal absorbance change, ΔAmax, when the binding equilibrium is forced to completion by mass action. The value of ΔAmax, in turn, allows calculation of the molar amount of cob(II)alamin species formed at the active site from the molar absorbtivity change upon complete conversion of Ado(L)Cbl to (L)cob(II)alamin (Δελ), and consequently the equilibrium constant for carbon–cobalt bond homolysis at the active site (Keq = kf/kr). The latter value then allows calculation of the individual rate constants since the observed rate constant for this equilibrium process is the sum of the forward and reverse rate constants (kobs = kf + kr). The results of this analysis are given in Table 1, along with those previously determined for AdoCbl itself.2
The results show that RTPR binds Ado(Bzim)Cbl slightly more weakly than it does AdoCbl, but binds Ado(Im)Cbl 8-fold more weakly. While the equilibrium constant for formation of the cobalt(II) species (Keq) is about the same for Ado(Bzim)Cbl and AdoCbl, it is about 5-fold smaller for Ado(Im)Cbl. The forward rate constant, kf, for enzymatic formation of the cobalt(II) species is nearly the same for Ado(Bzim)Cbl as for AdoCbl, but it is reduced by 17-fold for Ado(Im)Cbl. This suggests that the enzyme is less efficient at catalyzing the Co–C bond homolysis of Ado(Im)Cbl than that of AdoCbl. However, in order to determine if the steric bulk of the axial ligand plays a significant role in the enzymatic activation of the coenzyme, it is essential to know the intrinsic reactivity of the Co–C bond of Ado(Im)Cbl so that the catalytic efficiency of the enzyme with this analog can be determined and compared to that with AdoCbl.
Non-enzymatic thermolysis of Ado(Im)Cbl
In order to determine the catalytic efficiency of the enzyme with Ado(Im)Cbl, the kinetics of Ado(Im)Cbl non-enzymatic thermolysis were studied in anaerobic aqueous solution in the presence of a 50-fold molar excess of the radical trap 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (H-TEMPO) both spectrophotometrically (90–125 °C) and by a radiometric method (using [A2–3H]Ado(Im)Cbl at 105, 70, and 50 °C, and the method of initial rates for the lower two temperatures, as described previously3).1 Thermolysis of AdoCbl is well known to proceed via competing, parallel homolytic and heterolytic pathways (Scheme 2).3,34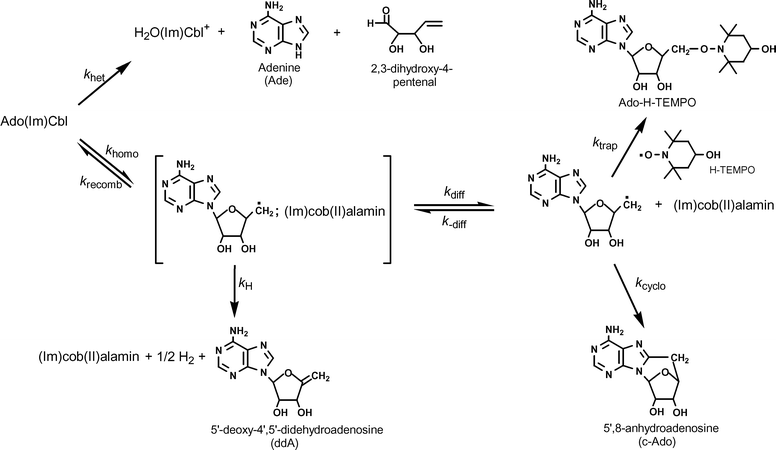 | ||
| Scheme 2 | ||
As is the case for all organocobalt compounds with β oxygen substituents,3 AdoCbl is acid labile, and undergoes a heterolytic process which yields aquacobalamin (H2OCbl+), adenine, and 2,3-dihydroxy-4-pentenal, in a process that is both specific acid-catalyzed and general acid (solvent) catalyzed.3,34,75,76 The competing process is carbon–cobalt bond homolysis, which is known to proceed through a solvent-caged cob(II)alamin-Ado˙ radical pair and consequently to be diffusion controlled in solution.34,51,77,78,79 The fate of the caged radical pair varies with conditions. An in-cage electron transfer process leads to 4′,5′-didehydroadenosine and hydrogen.78 Diffusive separation of the caged pair leads to a free Ado˙ radical which can undergo cyclization to 5′,8-anhydroadenosine,34,80,81 or, in the presence of a suitable radical trap such as H-TEMPO, form the trapped radical, Ado-H-TEMPO (Scheme 2). Under our conditions, with a large molar excess (50-fold) of radical trap, the only adenine containing products observed were adenine (from heterolysis) and Ado-H-TEMPO (from homolysis). The ratio of these products, then, provides a convenient method for determining the relative prevalence of the two competing pathways. The product ratio was measured1 at six temperatures between 70 and 125 °C and used to deconvolute the observed rate constants for thermolysis since for parallel first-order processes (Scheme 2), the observed rate constant is the sum of the rate constants for the parallel processes (eqn (2)), and the ratio of the products at any time is equal to the ratio of the rate constants (eqn (3)). A plot (Fig. 5) of the natural logarithm of the product ratio vs. 1/T was linear, as expected,3 since ln(khomo,obs/khet,obs) = [(ΔS‡homo,obs − ΔS‡het,obs)/R] − [(ΔH‡homo,obs − ΔH‡het,obs)/RT].
| kobs = khomo,obs + khet,obs | (2) |
| [IsoAdo-H-TEMPO]/[adenine] = khomo,obs/khet,obs | (3) |
 | (4) |
 | (5) |
 | (6) |
| Kbase-off = (1 + KCo)KBz | (7) |
![Plot of the natural logarithm of the ratio, R, of [Ado-H-TEMPO]/[Ade] observed for the thermolysis of Ado(Im)Cbl vs. 1/T. The solid line is a weighted least squares fit, slope = −8,099 ± 57, intercept = 21.57 ± 0.16, r2 = 0.998.](/image/article/2006/DT/b517599m/b517599m-f5.gif) | ||
| Fig. 5 Plot of the natural logarithm of the ratio, R, of [Ado-H-TEMPO]/[Ade] observed for the thermolysis of Ado(Im)Cbl vs. 1/T. The solid line is a weighted least squares fit, slope = −8,099 ± 57, intercept = 21.57 ± 0.16, r2 = 0.998. | ||
 | ||
| Scheme 3 | ||
Since significant amounts of the base-off species exist in solution even at neutral pH, the observed rate constants for the heterolytic and homolytic decomposition pathways are actually composites of the fundamental rate constants for the base-on and base-off species as seen in eqn (8) and (9), where αon is the fraction of Ado(Im)Cbl as the base-on species at any given temperature.
| khomo,obs = αonkhomo,on + (1 − αon)khomo,off | (8) |
| khet,obs = αonkhet,on + (1 − αon)khet,off | (9) |
 | ||
Fig. 6 Eyring plots for the homolysis (khomo,on, ●) and heterolysis (khet,on, ■) of Ado(Im)Cbl in water. The solid lines are least squares regressions: khomo,on, slope = −17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 ± 170 K, intercept = 6.91 ± 0.45, r2 = 0.999; khet,on, slope = −9,390 ± 170 K, intercept = −14.7 ± 0.5, r2 = 0.997. 500 ± 170 K, intercept = 6.91 ± 0.45, r2 = 0.999; khet,on, slope = −9,390 ± 170 K, intercept = −14.7 ± 0.5, r2 = 0.997. | ||
The comparison of the non-enzymatic thermolysis of Ado(Im)Cbl with that of AdoCbl (Table 2) is quite interesting. As was the case for AdoCbl,3 homolysis of Ado(Im)Cbl predominates at high temperatures, while heterolysis predominates at lower temperatures, the isokinetic point being 102.4°. At 100 °C, homolysis of Ado(Im)Cbl is 3.3-fold slower than homolysis of AdoCbl, and at 37 °C, Ado(Im)Cbl homolysis is 4.3-fold slower, the result of a 1.0 ± 0.4 kcal mol−1 increase in the enthalpy of activation for homolysis of Ado(Im)Cbl. The entropies of activation, in contrast, are identical. The situation for heterolysis is the reverse. Ado(Im)Cbl heterolysis is about 8.5-fold faster than AdoCbl heterolysis, regardless of the temperature, the result of identical enthalpies of activation, but an entropy of activation that is 4.9 ± 1.1 eu less negative than that for AdoCbl. As a result, at 100 °C, thermolysis of Ado(Im)Cbl proceeds with 54% heterolysis, compared to 4% heterolysis for AdoCbl at the same temperature.3,34 This is the result of a 3-fold decrease in the rate constant for homolysis of Ado(Im)Cbl coupled with a 9.4-fold increase in the rate constant for heterolysis. We attribute the decrease in the homolysis rate constant to the reduced corrin ring fold angle in Ado(Im)Cbl,1 consistent with the molecular modeling of mechanochemical triggering,59 and note that the effect of the steric bulk of the axial nucleotide is quite small. While there is a significant difference in basicity of the Im (pKa = 6.54 at 25 °C)1 and Bzm (pKa = 5.56 at 25 °C)97 axial nucleosides, we suspect that axial ligand basicity does not significantly affect Co–C bond homolysis as the reaction proceeds without the generation of any charge separation. In contrast, we suspect that the increase in heterolysis rate constant for Ado(Im)Cbl is the direct result of the increased basicity of the axial nucleoside. In this case, the reaction does produce a separation of charge (Scheme 4), and the emerging positive charge on the cobalt atom should be stabilized by increased basicity of the axial ligand. This is a conjecture which must be verified experimentally.
 | ||
| Scheme 4 | ||
Catalytic efficiency of RTPR with Ado(Im)Cbl
Extrapolation of the homolysis rate constant for Ado(Im)Cbl (Fig. 6) to 37 °C (Table 2) permits calculation of the catalytic efficiency of the enzyme with this coenzyme analog since the rate constant for enzyme-induced homolysis has also been measured (Table 1). Thus, while the enzyme-induced homolysis of Ado(Im)Cbl is 17-fold slower than that for AdoCbl, the non-enzymatic homolysis is also slower, but only by 4.3-fold. As a result, the catalytic efficiency of the enzyme for the homolysis of the Co–C bond of Ado(Im)Cbl (4 × 108, 12.2 kcal mol−1) is 3.8-fold lower than that for AdoCbl (1.5 × 109, 13.0 kcal mol−1),2 a difference of only 0.8 kcal mol−1. If our suspicion that axial ligand basicity does not significantly affect the homolysis rate is correct, then the steric bulk of the axial ligand contributes less than one kcal mol−1 to the enzymatic activation of AdoCbl, and classical mechanochemical triggering cannot be an important contributor to the 13 kcal mol−1 lowering of the activation energy for Co–C bond homolysis of AdoCbl generated by the RTPR enzyme.An experimental approach to product stabilization by axial Co–N bond length manipulation
If our conjecture (vide supra) is correct and axial Co–N bonds in Cbl's are abnormally long compared to simpler cobalt complexes due to steric interactions, then the axial Co–N orbital overlap in Cbl's is non-ideal. This suggests that an enzyme could electronically stabilize the cob(II)alamin product of Co–C bond homolysis by manipulation of the axial Co–N bond length so as to improve the orbital overlap. This, in turn, would require that the kinetics of cob(II)alamin formation at the enzyme active site be sensitive to axial ligand basicity, while chemical intuition (vide supra) suggests that non-enzymatic Co–C bond homolysis should be relatively insensitive to axial ligand basicity. It is thus important to determine the effect, if any, of axial ligand basicity on both the enzymatic and non-enzymatic Co–C bond homolysis of AdoCbl. To this end, we have used “guided biosynthesis” to obtain the cyano derivative of the cobalamin analog with 5-fluoroimidazole as the axial ligand (Fig. 1). As the pKa of 5-F–Im (2.4)98 is nearly five units below that of Im (7.2),99 direct comparison of our results with Ado(Im)Cbl to those with Ado(5-F–Im)Cbl should easily determine the effect of basicity on both enzymatic and non-enzymatic Co–C bond homolysis.Fermentation of P. shermanii on media supplemented with 4(5)-fluoroimidazole produced two cyanocobalamin analogs, one with a 4-fluoroimidazole axial nucleotide (CN(4-F–Im)Cbl) and one with a 5-fluoroimidazole axial ligand (CN(5-F–Im)Cbl). These were readily distinguishable by the prominent 1H NMR NOE crosspeak between the imidazole 5-H and the ribose 1-H in CN(4-F–Im)Cbl which is absent in the ROESY spectrum of CN(5-F–Im)Cbl. The latter has now been converted to Ado(5-F–Im)Cbl and characterized by 1H and 13C NMR and mass spectrometry.
A preliminary measurement of the kinetics of activation of Ado(5-F–Im)Cbl by RTPR has now been made. At 37 °C, kobs (kobs = kf + kr, eqn (1)) for (5-F–Im)Cbl is 0.12 s−1, fully 65-fold smaller than the value (kobs = 7.9 s−1, Table 1) for Ado(Im)Cbl. This striking result suggests that the activation of the coenzyme by RTPR is strongly dependent on the basicity of the axial ligand. Much work remains to be done here, including determination of the equilibrium constant for enzyme-induced homolysis to permit the separation of the forward and reverse rate constants, and measurement of the non-enzymatic thermal homolysis of Ado(5-F–Im)Cbl to determine if our suspicion that the non-enzymatic process will be relatively insensitive to axial ligand basicity is correct. However, if the implication of the preliminary observation proves correct, an important clue to the mechanism of enzymatic activation of AdoCbl will have been uncovered.
Acknowledgements
The author's research in this area is supported by the National Institute of General Medical Sciences (USA), Grant 48858.References
- K. L. Brown, X. Zou, R. R. Banka, C. B. Perry and H. M. Marques, Inorg. Chem., 2004, 43, 8130 CrossRef CAS.
- K. L. Brown and J. Li, J. Am. Chem. Soc., 1998, 120, 9466 CrossRef CAS.
- K. L. Brown and X. Zou, J. Inorg. Biochem., 1999, 77, 185 CrossRef CAS.
- S. Gschösser, R. B. Hannak, R. Konrat, K. Gruber, C. Mikl, C. Kratky and B. Kräutler, Chem. Eur. J., 2005, 11, 81 CrossRef.
- A. J. Brooks, M. Vlasie, R. Banerjee and T. C. Brunold, J. Am. Chem. Soc., 2004, 126, 8167 CrossRef CAS.
- B. T. Golding and W. Buckel, in Comprehensive Biological Catalysis, ed. M. L. Sinnott, Academic Press, London, 1997, p. 239 Search PubMed.
- R. Banerjee, Chem. Rev., 2003, 103, 2083 CrossRef CAS.
- K. Gruber and C. Kratky, Curr. Opin. Chem. Biol., 2002, 6, 598 CrossRef CAS.
- W. Buckel, G. Bröker, H. Bothe, A. J. Pierik and B. T. Golding, in Chemistry and Biochemistry of Vitamin B12, ed. R. Banerjee, Wiley, New York, 1999, p. 757 Search PubMed.
- K. Zerbe-Burkhardt, A. Ratnatilleke, J. W. Vrijbloed and J. A. Robinson, in Chemistry and Biochemistry of Vitamin B12, ed. R. Banerjee, Wiley, New York, 1999, p. 859 Search PubMed.
- T. Toraya, Chem. Rec., 2002, 2, 352 CrossRef CAS.
- T. Toraya, Cell. Mol. Life Sci., 2000, 57, 106 CrossRef CAS.
- V. Bandarian and G. H. Reed, in Chemistry and Biochemistry of Vitamin B12, ed. R. Banerjee, Wiley, New York, 1999, p. 811 Search PubMed.
- S. Licht and J. Stubbe, in Comprehensive Natural Product Chemistry, ed. C. D. Poulter, Elsevier, Amsterdam, 1999, vol. 5, p. 163 Search PubMed.
- P. A. Frey and C. H. Chang, in Chemistry and Biochemistry of Vitamin B12, ed. R. Banerjee, Wiley, New York, 1999, p. 835 Search PubMed.
- G. H. Reed and S. O. Mansoorabadi, Curr. Opin. Struct. Biol., 2003, 13, 716 CrossRef CAS.
- R. Padmakumar, S. Taoke, R. Padmakumar and R. Banerjee, J. Am. Chem. Soc., 1995, 117, 7033 CrossRef CAS.
- O. Zelder, B. Beatrix, F. Kroll and W. Buckel, FEBS Lett., 1995, 369, 252 CrossRef CAS.
- F. Mancia, N. H. Keep, A. Nakagawa, P. F. Leadley, S. McSweeney, B. Rasmussen, P. Bösecke, O. Diat and P. R. Evans, Structure, 1996, 4, 339 CrossRef CAS.
- R. Reitzer, K. Gruber, G. Jogl, U. G. Wagner, H. Bothe, W. Buckel and C. Kratky, Structure, 1999, 7, 891 CrossRef.
- M. Yamanishi, S. Yamada, H. Muguruma, Y. Murakami, T. Tobimatsu, A. Ishida, J. Yamauchi and T. Toraya, Biochemistry, 1998, 37, 4799 CrossRef CAS.
- C. C. Lawrence, G. J. Gerfen, V. Samano, R. Nitsche, J. J. Robins, J. Rétey and J. Stubbe, J. Biol. Chem., 1999, 274, 7039 CrossRef CAS.
- A. Abend, V. Bandarian, R. Nitsche, E. Stupperich, J. Rétey and G. H. Reed, Arch. Biochem. Biophys., 1999, 370, 138 CrossRef CAS.
- N. Shibata, J. Masuda, T. Tobimatsu, T. Toraya, K. Suto, Y. Morimoto and N. Yasuoka, Structure, 1999, 7, 997 CrossRef CAS.
- M. D. Sinchak, G. Arjara, B. A. Kellogg, J. Stubbe and C. L. Drennan, Nat. Struct. Biol., 2002, 9, 293 CrossRef CAS.
- C. H. Chang and P. A. Frey, J. Biol. Chem., 2000, 275, 106 CrossRef CAS.
- H.-P. Chen, S.-H. Wu, Y.-L. Lin, C.-M. Chen and S.-S. Tsay, J. Biol. Chem., 2001, 276, 44744 CrossRef CAS.
- K. L. Brown, X. Zou, G. Chen, Z. Xia and H. M. Marques, J. Inorg. Biochem., 2004, 98, 287 CrossRef CAS.
- T. Toraya, in Vitamin B12 and B12-Proteins, ed. B. Kräutler, D. Arigoni and B. T. Golding, Wiley, VCH, Weinheim, 1998, p. 303 Search PubMed.
- M. I. Yakusheva, A. A. Poznanskaya, T. A. Pospelova, I. P. Rudakova, A. Yurkevich and V. A. Yakolev, Biochim. Biophys. Acta, 1977, 484, 216 CrossRef CAS.
- H. Bothe, G. Böker, U. Müller, I. Schall, S. Textor, B. T. Golding and W. Buckel, in Vitamin B12 and B12-Proteins, ed. B. Kräutler, D. Arigoni and B. T. Golding, Wiley VHC, Weinheim, 1998, p. 237 Search PubMed.
- S. Chowdhury, M. G. Thomas, J. C. Escalante-Semerena and R. Banerjee, J. Biol. Chem., 2001, 276, 1015 CrossRef CAS.
- U. Weigl, M. Heimberger, A. J. Pierik and J. Rétey, Chem. Eur. J., 2003, 9, 652 CrossRef CAS.
- B. P. Hay and R. G. Finke, J. Am. Chem. Soc., 1986, 108, 4820 CrossRef CAS.
- T. Koenig and R. G. Finke, J. Am. Chem. Soc., 1988, 110, 2657 CrossRef CAS.
- K. L. Brown and L. Zhou, Inorg. Chem., 1996, 35, 5032 CrossRef CAS.
- S. B. Booker and J. Stubbe, Proc. Natl. Acad. Sci. USA, 1993, 90, 8352 CAS.
- R. L. Blakley, in B12, vol. 2, ed. D. Dolphin, Wiley, New York, 1982, p. 381 Search PubMed.
- H. P. C. Hogenkamp, R. K. Ghambeer, C. Bronson and R. L. Blakley, Biochem. J., 1967, 103, 5C CAS.
- W. H. Orme-Johnson, H. Beinert and R. L. Blakley, J. Biol. Chem., 1974, 249, 2338 CAS.
- S. Licht, G. J. Gerfen and J. Stubbe, Science, 1996, 271, 477 CrossRef CAS.
- T. Tomao and R. L. Blakley, Biochemistry, 1973, 12, 24 CrossRef.
- S. S. Licht, C. C. Lawrence and J. Stubbe, Biochemistry, 1999, 38, 1234 CrossRef CAS.
- S. Booker, S. Licht, J. Broderick and J. Stubbe, Biochemistry, 1994, 33, 12676 CrossRef CAS.
- D. Chen, A. Abend, J. Stubbe and P. A. Frey, Biochemistry, 2003, 42, 4578 CrossRef CAS.
- J. H. Grate and G. N. Schrauzer, J. Am. Chem. Soc., 1979, 101, 4601 CrossRef CAS.
- S. M. Chamaly and J. M. Pratt, J. Chem. Soc., Dalton Trans., 1980, 2274 RSC.
- J. Halpern, Bull. Soc. Chim. Fr., 1988, 187 CAS.
- V. B. Pett, M. N. Liebman, P. Murray-Rust, K. Prasad and J. P. Glusker, J. Am. Chem. Soc., 1987, 109, 3207 CrossRef CAS.
- N. Bresciani-Pahor, M. Forcolin, L. G. Marzilli, L. Randaccio, M. F. Summers and P. J. Toscano, Coord. Chem. Rev., 1985, 63, 1 CrossRef CAS.
- B. P. Hay and R. G. Finke, J. Am. Chem. Soc., 1987, 109, 8012 CrossRef CAS.
- T. Toraya and A. Ishida, Biochemistry, 1979, 18, 417 CrossRef CAS.
- K. L. Brown and H. B. Brooks, Inorg. Chem., 1991, 30, 3420 CrossRef CAS.
- H. F. J. Savage, P. F. Linndley, J. L. Finney and P. A. Timmins, Acta Crystallogr., Sect. B, 1987, 43, 280 CrossRef.
- M. F. Summers, P. J. Toscano, N. Bresciani-Pahr, G. Nardin, L. Randaccio and L. G. Marzilli, J. Am. Chem. Soc., 1983, 105, 6259 CrossRef CAS.
- K. L. Brown and H. M. Marques, J. Mol. Struct. (THEOCHEM), 2005, 714, 209 CrossRef CAS.
- L. Randaccio, M. Furlan, S. Geremia, M. Slouf, I. Srnova and D. Toffoli, Inorg. Chem., 2000, 39, 3403 CrossRef CAS.
- J.-P. Charland, E. Zangrando, N. Bresciani-Pahor, L. Randaccio and L. G. Marzille, Inorg. Chem., 1993, 32, 4256 CrossRef.
- K. L. Brown and H. M. Marques, J. Inorg. Biochem., 2001, 83, 121 CrossRef CAS.
- J. M. Sirovatka, J. M. Rappé and R. G. Finke, Inorg. Chim. Acta, 3000, 300, 545.
- C. Kratky, G. Färber, K. Gruber, Z. Deuter, H. F. Nolting, R. Konrat and B. Kräutler, J. Am. Chem. Soc., 1995, 117, 4654 CrossRef CAS.
- A. Marchaj, D. W. Jacobsen, S. R. Savon and K. L. Brown, J. Am. Chem. Soc., 1995, 117, 11640 CrossRef CAS.
- H. M. Marques and K. L. Brown, Inorg. Chem., 1995, 34, 3733 CrossRef CAS.
- B. Kräutler, R. Konrat, E. Stupperich, G. Färber, K. Gruber and C. Kratky, Inorg. Chem., 1994, 33, 4128 CrossRef.
- E. Stupperich, I. Steiner and M. Rühlemann, Anal. Biochem., 1986, 155, 365 CAS.
- J. A. Horig, P. Renz and G. Heckmann, J. Biol. Chem., 1978, 253, 7410 CAS.
- P. Renz, Methods Enzymol., 1971, 18C, 82.
- K. L. Brown, X. Zou, J. Li and G. Chen, Inorg. Chem., 2001, 40, 5942 CrossRef CAS.
- C. Schöneich and K.-D. Asmus, Radiat. Environ. Biophys., 1990, 29, 263 CAS.
- C. Schöneich, K.-D. Asmus and M. Bonifacic, J. Chem. Soc., Faraday Trans., 1995, 91, 1923 RSC.
- W. Karmann, A. Granzow, G. Meissner and A. Henglein, Int. J. Radiat. Phys. Chem., 1969, 1, 395 Search PubMed.
- J. Redpath, Radiat. Res., 1973, 54, 364 CrossRef CAS.
- J. A. Franz, B. A. Bushaw and M. S. Alnajjar, J. Am. Chem. Soc., 1989, 111, 268 CrossRef CAS.
- P. Huston, J. H. Espenson and A. Bakac, Inorg. Chem., 1992, 31, 720 CrossRef.
- L. E. H. Gerards and S. Balt, Recl. Trav. Chim. Pays-Bas, 1992, 111, 411 CAS.
- L. E. H. Gerards and S. Balt, Recl. Trav. Chim. Pays-Bas, 1994, 113, 137 CAS.
- B. P. Hay and R. G. Finke, Polyhedron, 1988, 7, 1469 CrossRef CAS.
- C. D. Garr and R. G. Finke, J. Am. Chem. Soc., 1992, 114, 10440 CrossRef CAS.
- C. D. Garr and R. G. Finke, Inorg. Chem., 1993, 32, 4414 CrossRef.
- H. P. C. Hogenkamp, J. Biol. Chem., 1963, 238, 477 CAS.
- K. N. V. Duong, A. Gaudemer, M. D. Johnson, R. Quillivic and J. Zyber, Tetrahedron Lett., 1975, 34, 2997 CrossRef.
- K. L. Brown and S. Peck-Siler, Inorg. Chem., 1988, 27, 3548 CrossRef CAS.
- K. L. Brown and G.-Z. Wu, Inorg. Chem., 1994, 33, 4122 CrossRef CAS.
- K. L. Brown, J. M. Hakimi, D. M. Nuss, Y. D. Mantejano and D. W. Jacobsen, Inorg. Chem., 1984, 23, 1463 CrossRef CAS.
- P. A. Levene and E. T. Stiller, J. Biol. Chem., 1933, 102, 187 CAS.
- R. S. Klein, H. Ohrui and J. J. Fox, J. Carbohydr., Nucleosides, Nucleotides, 1974, 1, 265 Search PubMed.
- T. Mukaiyama, Y. Hashimoto, Y. Hayashi and S.-I. Shoda, Chem. Lett., 1984, 557 CAS.
- S. DeBarnardo and M. Weigele, J. Org. Chem., 1976, 41, 287 CrossRef.
- R. J. Blau and J. H. Espenson, J. Am. Chem. Soc., 1985, 107, 3530 CrossRef CAS.
- J. D. Brodie, Proc. Natl. Acad. Sci. USA, 1969, 62, 461 CAS.
- J. H. Grate and G. N. Schrauzer, J. Am. Chem. Soc., 1979, 101, 4601 CrossRef CAS.
- K. L. Brown and H. B. Brooks, Inorg. Chem., 1991, 30, 3420 CrossRef CAS.
- M. D. Waddington and R. G. Finke, J. Am. Chem. Soc., 1993, 115, 4629 CrossRef CAS.
- K. L. Brown, S. Cheng and H. M. Marques, Inorg. Chem., 1995, 34, 3038 CrossRef CAS.
- K. L. Brown, L. Salmon and J. A. Kirby, Organometallics, 1992, 11, 422 CrossRef CAS.
- M. P. Jensen and J. Halpern, J. Am. Chem. Soc., 1999, 121, 2181 CrossRef CAS.
- K. L. Brown, J. M. Hakimi and D. W. Jacobsen, J. Am. Chem. Soc., 1984, 106, 7894 CrossRef CAS.
- H. J. C. Yeh, K. L. Kirk and L. A. Cohen, J. Chem. Soc., Perkin Trans. 2, 1975, 928 RSC.
- H. M. Marques, T. J. Egan, J. H. Marsh, J. R. Mellor and O. Q. Munro, Inorg. Chim. Acta, 1989, 166, 249 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |