Mechanochemical preparation of molecular and supramolecular organometallic materials and coordination networks
Dario
Braga
*,
Stefano L.
Giaffreda
,
Fabrizia
Grepioni
,
Anna
Pettersen
,
Lucia
Maini
,
Marco
Curzi
and
Marco
Polito
Dipartimento di Chimica G. Ciamician, Università degli Studi di Bologna, Via F. Selmi 2, 40126, Bologna, Italy. E-mail: dario.braga@unibo.it
First published on 10th February 2006
Abstract
This Dalton Perspective deals with solvent-free reactions taking place within solids or between solids or involving a solid and a vapour. The focus is on reactions involving organometallic and coordination compounds and occurring via reassembling of non-covalent bonding, e.g. hydrogen bonds, and/or formation of ligand–metal coordination bonds. It is argued that reactions activated by mechanical mixing of solid reactants as well as those obtained by exposing a crystalline solid to a vapour can be exploited to “make crystals”, which is the quintessence of crystal engineering. It is demonstrated through a number of examples that solvent-free methods, such as co-grinding, kneading, milling of molecular solids, or reactions of solid with vapours represent viable alternative, when not unique, routes for the preparation of novel molecular and supramolecular solids as well as for the preparation of polymorphic or solvate modifications of a same species. The structural characterization of the products requires the preparation of single crystals suitable for X-ray diffraction, a goal often achieved by seeding.
 Dario Braga’s research group | Dario Braga is Professor of Chemistry at the University of Bologna. Presently, his main scientific interests are in the study of crystal polymorphism and in the crystal engineering exploitation of hydrogen-bonding interaction between ions and in solvent-free gas–solid and solid–solid reactions. He received the Raffaello Nasini Prize from the Italian Society of Chemistry in 1988 for his studies on solid-state dynamic processes, and the FEDERCHIMICA Prize for 1995 for his research on the intermolecular interactions in organometallic systems. He has published more than 300 papers and reviews and organized several international conferences and schools on crystal engineering. He is Scientific Editor of CrystEngComm, member of the international editorial board of Chem. Commun. member of the Institute of Advanced Study and Director of the Collegio Superiore of the Alma Mater Studiorum University of Bologna. |
Stefano L. Giaffreda was born in 1977 in Mesagne (BR), Italy. He graduated at the University of Bologna in 2003. Currently he is a PhD student in the group of Professor Dario Braga. His scientific interests are in the fields of organometallic synthesis and crystal engineering. |
Fabrizia Grepioni is Associate Professor of Structural and Functional Materials and of General Chemistry at the University of Bologna, after spending six years as Associate Professsor at the University of Sassari. She received a PhD from the University of Bologna in 1990. She was awarded the 1997 Raffaello Nasini Prize from the Italian Society of Chemistry for her studies on intermolecular interactions in organometallic solids. She has published more than 200 papers. Her scientific interests are in the fields of crystal engineering and molecular crystal polymorphism. |
Anna Pettersen (née Johansson) is a Post-doc in the research group of Professor Dario Braga and was born in 1977 in Karlskrona, Sweden. She graduated at Göteborg University, Sweden, in 2000, where she also received her PhD in organic chemistry in 2005 under the supervision of Professor Mikael Håkansson. She has received fellowships from the Bertil Lundqvist foundation as well as the Swedish Research Council for postdoctoral studies at Bologna University. Her scientific interests are in the fields of organometallic structural chemistry and solvent-free chemistry. |
Lucia Maini was born in 1972 in Bologna, Italy. During her undergraduate degree at the University of Bologna she carried out her undergraduate thesis at the University of California under the supervision of Prof. Peter C. Ford. She received her PhD degree in Chemistry at the University of Bologna under the direction of Prof. D. Braga. Currently she works as a postdoctoral research fellow with Prof. Braga. Her main interests are in solvent-free chemistry, crystal engineering and polymorphism. |
Marco Curzi was born in 1974 in Nereto (TE), Italy. He graduated at the University of Bologna in December 2003 and received a Spinner fellowship in 2005. He is now a member of PCL (PolyCrystalLine) and his main scientific interests are in polymorphism and solvent-free chemistry. |
Marco Polito was born in Gorizia (Italy) in 1974. He earned his PhD in Inorganic Chemistry at the University of Bologna in June 2004. Currently he has a postdoctoral position in Bologna in the laboratories of Prof. Dario Braga. His current research interests are related to organometallic chemistry, polymorphism and heterophase reactions. |
Introduction
The core paradigm of crystal engineering is the ability of assembling molecules or ions into periodical functional structures, the crystal, by controlling molecular recognition and aggregation via supramolecular interactions and coordination bonds.1–7 Since the product of a crystal engineering experiment is by definition in the crystalline form, crystal engineers are crystal makers who constantly (and inevitably) face the problem of obtaining crystals for the characterization of their reaction products. Whether the products are obtained from solution, melt, vapour or from more forcing hydrothermal syntheses they will have to be investigated and characterized by solid-state techniques, in particular by X-ray diffraction. Amorphous materials are also extremely interesting,8–16 but their investigation and use is still very limited mainly in view of the difficulty in their characterization.In this perspective article we will demonstrate that novel crystalline materials can be obtained by reacting preformed crystalline materials with solid-state reactants in “solvent-free” conditions.17 We will argue that “mechanochemical” reactions between solids represent alternative and, at times, unique ways to prepare a variety of crystalline materials from coordination networks to hydrogen bonded adducts etc.
It should be pointed out that part of the work discussed hereafter has been described previously in related review articles.18–20 This perspective, however, is focused on the utilization of metal complexes as starting materials, which has been traditionally the field of interest of our Bolognese crystal engineering laboratory.21–23
Making crystalline materials by mechanochemical methods
The formation of crystals as products in solvent-free reactions between solid materials is controlled by the possibility of bringing reactants into contact. Therefore, intimate mixing of the reactants and large surface areas (i.e. small particle size) are, generally speaking, a prerequisite for successful reaction. Reactions between polycrystalline powders are usually carried out by mechanochemical methods such as manual co-grinding or milling (vide infra). Such methods generally produce materials also in the form of a polycrystalline powder and are, rather obviously, not suited for the growth of single crystals (for single-crystal diffractometers). Since mechanochemical mixing of reactants ultimately amounts to “making crystals by smashing crystals” the single-crystal dogma, on which the vast majority of crystal engineering studies is based, is seemingly contradicted. Unless one recurs to high intensity synchrotron radiation, microcrystals will allow only powder diffraction experiments, which only rarely can be used for ab initio structure determination in order to get those precise structural information that are so essential to the crystal engineer. Full structural characterization of the products, however, still replies on the possibility of obtaining single crystals, which might be often grown from solutions of the desired product by the seeding technique. Single-crystal X-ray diffraction experiments, in fact, will not only allow to know the structure of the products in fine details but also to carry out a useful comparison between the powder diffractograms measured on the bulk product and those calculated on the basis of the single-crystal structures. This is widely applied in the cases discussed herein.Non-solution methods whether from solid–gas or solid–solid reactions (see Fig. 1) require the chemist, or crystal engineer, to explore/exploit methods that are not routinely used in chemical labs such as grinding and milling, which are less popular, when they are not dismissed as non-chemical, in academic research labs.24–27
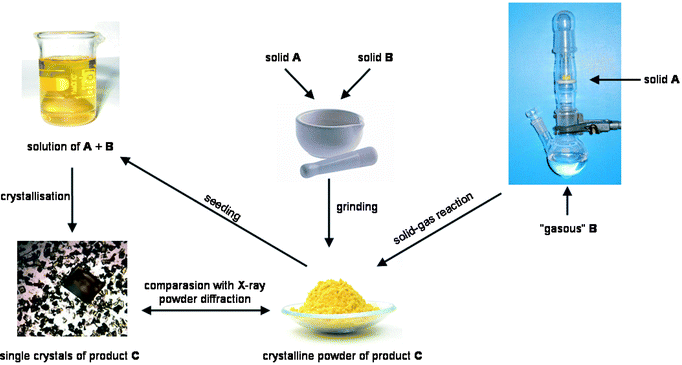 | ||
| Fig. 1 The solid–gas (right) and solid–solid (middle) processes and the strategy to obtain single crystals by recrystallisation of the solid reaction product in the presence of seeds (left) of the desired crystals. | ||
Grinding and milling
Typical mechanochemical reactions are those activated by co-grinding or milling of powder materials. These reactions are usually carried out either manually, in an agate mortar, or electro-mechanically, as in ball milling. In both cases the main difficulty is in controlling reaction conditions: grinding time, temperature, pressure exerted by the operator, etc. Furthermore, the heat generated in the course of the mechanochemical process can induce local melting of crystals or melting at the interface between the different crystals, so that the reaction might take place in the liquid phase even though solid products are ultimately recovered. One should also keep in mind that mechanical stress by fracturing the crystals increases surface area and facilitates interpenetration and reaction depending on the ability of molecules to diffuse through the crystal surfaces. Mechanochemical methods are commonly used at industrial level mainly with inorganic solids.28–31In some cases the use of a small quantity of solvent can accelerate (when not make altogether possible) solid-state reactions carried out by grinding or milling.32 The method based on the co-grinding of powdered reactants in the presence of a small amount of solvent, also known as kneading, has been described as a sort of “solvent catalysis” of the solid-state process, whereby the small amount of solvent provides a lubricant for molecular diffusion. The objection about whether a kneaded reaction between two solid phases can be regarded as a bona fide solid-state process is justified. However, in the context of this work, the interest lies more in the methods to make new crystalline materials rather than in the mechanisms. Industrial applications of kneading have been developed for pharmaceutical powders.33,34 As an example of preparative lab scale one could mention the preparation by kneading of binary β-cyclodextrin bifonazole,35 and of β-cyclodextrin inclusion compounds of ketoprofen,36 ketoconazole37 and carbaryl.38
We have pointed out that, even though the product of a grinding, milling and kneading process is, in general, in the form of a powdered material, single crystals are still highly desirable (when not indispensable) for a thorough characterization of the reaction product. In such a case, seeding can provide a route to the growth of single crystals of the desired material.17,39–41 The method, however, is not failure-proof, and other conditions, such as supersaturation, rate of solvent evaporation and temperature as well as the purity of the seeding powder obtained by grinding may still play a role in determining the crystallization outcome.
The use of seeds is also very important when there is the possibility of formation of different crystal forms, i.e. crystal polymorphism.42,43Seeds of isostructural or quasi-isostructural species that crystallise well can also been employed to induce crystallisation of unyielding materials (heteromolecular seeding).44–46 For instance, chiral co-crystals of tryptamine and hydrocinnamic acid have also been prepared by crystallization in the presence of seeds of different chiral crystals.47 Of course, unintentional seeding may also alter the crystallization process in an undesired manner.48
Mechanically-induced formation of covalent bonds
While mechanochemical methods have been widely used with organic26 and inorganic24 compounds, there are not many examples of the utilization of mechanochemical procedures in coordination chemistry. For instance, Balema et al. have shown that the cis-platinum complexes cis-(Ph3P)2PtCl2 and cis-(Ph3P)2PtCO3 can be prepared mechanochemically from solid reactants in the absence of solvent.49 Orita et al. have reported that the reaction of (ethylenediamine)Pt(NO3)2 with 4,4′-bipyridine, which takes as long as 4 weeks at 100 °C to form metallamacrocyclic molecular squares,50,51 is brought to completion within 10 min at room temperature by mixing reactants without solvents.52 Similar reaction acceleration has been observed also with triazine based ligands.53,54 Double helix formation under solvent free conditions has also been achieved by reacting chiral oligo(bipyridine) copper complexes with [(CH3CN)4Cu]PF6. The progress of the reaction was monitored by measuring solid-state CD-spectra showing that after grinding for 5 min the desired helicate had been obtained.52It has been reported that bis-substituted pyridine/pyrimidine ferrocenyl complexes can be prepared by mechanically-induced Sukuzi-coupling reaction55–59 in the solid-state starting from ferrocene-1,1′-diboronic acid, [Fe(η5-C5H4–B(OH)2)2] (see Scheme 1).60 The solvent-free reactions allow synthesis in the air and at room temperature of mono- and bis-substituted pyridine and pyrimidine ferrocenyl derivatives thus providing a valuable alternative to the preparation in solution. Actually, in the case of [Fe(η5-C5H4-1-C5H4N)2], the solvent-free process is much faster and more selective than the same reaction carried out in solution. Beside shorter reaction times, less workup, higher yield, and the absence of solvents the solid-state reaction affords the possibility of combining different synthetic steps in order to obtain homo- and hetero-ligands ferrocenyl complexes.
![The solid-state synthesis of mono- and bis-substituted pyridine and pyrimidine ferrocenyl derivatives starting from ferrocene-1,1′-diboronic acid, [Fe(η5-C5H4–B(OH)2)2].](/image/article/2006/DT/b516165g/b516165g-s1.gif) | ||
| Scheme 1 The solid-state synthesis of mono- and bis-substituted pyridine and pyrimidine ferrocenyl derivatives starting from ferrocene-1,1′-diboronic acid, [Fe(η5-C5H4–B(OH)2)2]. | ||
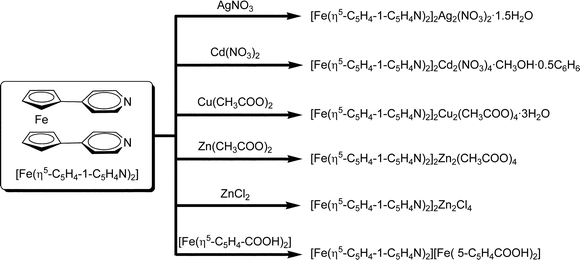
Preparation of “complexes-of-complexes”: mixed metal superstructures derived from bispyridine
The bidentate compounds described in the previous section were employed for the preparation of supramolecular “complexes-of-complexes”,61 aiming to the construction of mixed-metal supramolecular materials with interesting electrochemical and spin properties. The bottom-up construction of supramolecular materials and coordination networks with desired properties is one of the main goals of crystal engineering.62–91 Ferrocene-based pyridyl ligands have been studied for the possibility of exploiting their redox properties in various applications, such as amperometric sensors for metal ions. The focus, however, has been mainly on mono-substituted ferrocenes, while only few examples of bis-substituted ferrocene pyridyl complexes are known.92–99 We have prepared a series of mixed-metal macrocyclic complexes by reacting mechanochemically prepared [Fe(η5-C5H4-1-C5H4N)2] with transition metal salts, such as AgNO3, Cd(NO3)2, Cu(CH3COO)2, Zn(CH3COO)2 and ZnCl2 (Scheme 2).100 A similar approach has been used before by others by using aminocobaltocenes and aminoferrocenes to form complexes with Zn2+ and Co2+ metal ions.95,101–117 while flexible bis-p-aminopyridine bidentate ligands have also been utilized to prepare metallamacrocycle complexes.118–121 Analogously, neutral ligands (1,4-diazabicyclooctane, tetramethylpyrazine and pyrazine) have been used to link silver carboxylates in extended networks.122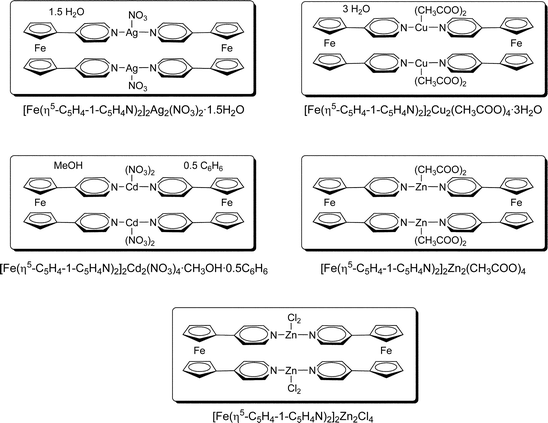 | ||
| Scheme 2 | ||
In view of the conformational freedom of the two η5-C5H4-1-C5H4N ligands in [Fe(η5-C5H4-1-C5H4N)2] we were also intrigued by the question on whether the ligands would have adopted a cisoid or a transoid conformation upon coordination.123 While the former conformation leads to the formation of finite coordination geometry, i.e. to a molecular complex, the latter might, in principle, lead to an infinite network. The two limiting situations are shown in Fig. 2.
![The cisoid and transoid conformation of two metallamacrocycle compounds (a) the [Fe(η5-C5H4-4-C5H4N)2. The [Fe(η5-C5H4-4-C5H4N)2] leads to a finite structure (as in (b)) but it could also, in principle, form a infinite network (as in (c)), even though the structure in (c) is a fictive one.](/image/article/2006/DT/b516165g/b516165g-f2.gif) | ||
| Fig. 2 The cisoid and transoid conformation of two metallamacrocycle compounds (a) the [Fe(η5-C5H4-4-C5H4N)2. The [Fe(η5-C5H4-4-C5H4N)2] leads to a finite structure (as in (b)) but it could also, in principle, form a infinite network (as in (c)), even though the structure in (c) is a fictive one. | ||
However, all compounds in Scheme 3 do not form extended networks but metallamacrocycles. The solid-state structure of the heterobimetallic system complex [Fe(η5-C5H4-1-C5H4N)2]2Ag2(NO3)2·1.5H2O is shown in Fig. 3. The two silver atoms interact directly with the pyridine ligands, with Ag⋯N distances between 2.123(8), and 2.145(8) Å. The Ag⋯Ag distance is 3.500(2) Å. This separation is comparable to that observed in other dimeric Ag-complexes.124,125 The two nitrate anions play different roles in the crystal structure. While one is directly linked to the other nitrate anion via water molecules, the second anion acts as a bridge between dimeric units as shown in Fig. 4, with O⋯Ag distances of 2.752(9) and 2.870(1) Å, respectively. A similar ion-pairing link between neighbouring complexes has been observed in the complex with Ag+ and bis-p-aminopyridine bidentate ligands.
 | ||
| Scheme 3 | ||
![The heterometallic [Fe(η5-C5H4-1-C5H4N)2]2Ag2(NO3)2·1.5H2O complex showing how the how the network is built up by bridging nitrate anions between the dimeric units.](/image/article/2006/DT/b516165g/b516165g-f3.gif) | ||
| Fig. 3 The heterometallic [Fe(η5-C5H4-1-C5H4N)2]2Ag2(NO3)2·1.5H2O complex showing how the how the network is built up by bridging nitrate anions between the dimeric units. | ||
![(a) The crystal structure of the [Fe(η5-C5H4-1-C5H4N)2]2Zn2(CH3COO)4 complex. (b) The space filling model of the [Fe(η5-C5H4-1-C5H4N)2]2Zn2(CH3COO)4 complex showing the packing.](/image/article/2006/DT/b516165g/b516165g-f4.gif) | ||
| Fig. 4 (a) The crystal structure of the [Fe(η5-C5H4-1-C5H4N)2]2Zn2(CH3COO)4 complex. (b) The space filling model of the [Fe(η5-C5H4-1-C5H4N)2]2Zn2(CH3COO)4 complex showing the packing. | ||
The complex [Fe(η5-C5H4-1-C5H4N)2]2Zn2(CH3COO)4 shows that the Cp-pyridine ligands are nearly eclipsed with a Zn⋯Zn separation of 3.875(1) Å. The two acetate ligands span the Zn⋯Zn system [Zn(1)–O(3) 2.018(3), Zn(1)–O(4) 2.018(2) Å] while two acetate ligands are dangling externally at a Zn⋯O separation of 2.042(3) Å (see Fig. 4). The Zn⋯Zn distance indicates that the interaction between the Zn centres is entirely repulsive, as would be expected from closed shell first row systems. The coordination around the Zn-centres can be described as a trigonal bipyramid. A space filling representation of the packing of [Fe(η5-C5H4-1-C5H4N)2]2Zn2(CH3COO)4 in the solid state is shown in Fig. 4(b).
The structure of the compound obtained by reacting the bispyridine with zinc chloride, [Fe(η5-C5H4-1-C5H4N)2]2Zn2Cl4 is shown in Fig. 5. The constrain imposed by the tetrahedral coordination around the zinc centres [Zn–Cl distances 2.202(3), 2.224(3) Å] leads to the formation of a butterfly-type molecule, with the two ZnCl2 units forming the hinge and the ferrocenyl units the wings of the butterfly Å (see Fig. 6(b) for a space filling representation). The distance between the two Zn centres is 6.125 Å.
![(a) The crystal structure of [Fe(η5-C5H4-1-C5H4N)2]2Zn2Cl4 showing tetrahedral coordination geometry around the zinc centres. (b) This coordination geometry will lead to the formation of a butterfly-type molecule as shown by the space filling representation.](/image/article/2006/DT/b516165g/b516165g-f5.gif) | ||
| Fig. 5 (a) The crystal structure of [Fe(η5-C5H4-1-C5H4N)2]2Zn2Cl4 showing tetrahedral coordination geometry around the zinc centres. (b) This coordination geometry will lead to the formation of a butterfly-type molecule as shown by the space filling representation. | ||
![(a) The crystal structure of the [Fe(η5-C5H4-1-C5H4N)2]2Cd2(NO3)4·CH3OH·0.5C6H6 complex showing the coordinated methanol molecule which leads to different coordination numbers around the two cadmium centres. (b) The complex form ample channels throughout the structure where solvent molecules (benzene) can be accommodated.](/image/article/2006/DT/b516165g/b516165g-f6.gif) | ||
| Fig. 6 (a) The crystal structure of the [Fe(η5-C5H4-1-C5H4N)2]2Cd2(NO3)4·CH3OH·0.5C6H6 complex showing the coordinated methanol molecule which leads to different coordination numbers around the two cadmium centres. (b) The complex form ample channels throughout the structure where solvent molecules (benzene) can be accommodated. | ||
The structure of [Fe(η5-C5H4-1-C5H4N)2]2Cd2(NO3)4·CH3OH·0.5C6H6 is reminiscent of that of the silver complex with the notable difference that the Cd-centres are bridged by two asymmetric nitrate bridges [Cd(1)–O(9) 2.355(7), Cd(2)–O(9) 2.400(6), Cd(1)–O(1) 2.450(6), Cd(2)–O(1) 2.757(6) Å]. While the Cd(1) centre is six-coordinated, the Cd(2) centre is seven-coordinated because of additional presence of a coordinated methanol molecule (see Fig. 6). In summary, one Cd-centre is almost octahedral, while the other is seven-coordinated and the Cd⋯Cd distance is 4.225(3) Å. The ferrocenyl-pyridine systems stack at a distance of 3.47 Å, with the Cd-atoms completely screened from the surrounding by the N- and O-interactions. Fig. 6 shows how the supramolecular arrangement of the complexes leaves ample channels in the structure where the solvent molecules (light blue) can be accommodated.
The complex [Fe(η5-C5H4-1-C5H4N)2]2Cu2(CH3COO)4·3H2O possesses a structure that is reminiscent of that of [Fe(η5-C5H4-1-C5H4N)2]2Ag2(NO3)2·1.5H2O, the Cp-pyridine ligands are in eclipsed conformation and bridge the two Cu-atoms (see Fig. 7). The Cu⋯Cu separations are 3.428(5) and 3.473(5) Å, respectively, for the two independent half molecules in the asymmetric unit. Similarly to what observed in [Fe(η5-C5H4-1-C5H4N)2]2Cd2(NO3)4·CH3OH·0.5C6H6 the bimetallic Cu⋯Cu unit is spanned by two acetate anions [Cu(1)–O(2) 1.96(2), 2.49(2), Cu(2)–O(6) 2.06(2), 2.37(2) Å]. If one considers the additional acetate ions linked to each Cu atom, the coordination around copper can be described as distorted square pyramidal.
![The heterometallic [Fe(η5-C5H4-1-C5H4N)2]2Cu2(CH3COO)4·3H2O complex showing how the how the network is built up by bridging acetate anions between the dimeric units.](/image/article/2006/DT/b516165g/b516165g-f7.gif) | ||
| Fig. 7 The heterometallic [Fe(η5-C5H4-1-C5H4N)2]2Cu2(CH3COO)4·3H2O complex showing how the how the network is built up by bridging acetate anions between the dimeric units. | ||
Clearly the complex [Fe(η5-C5H4-1-C5H4N)2] can also take part in hydrogen-bonding interactions with two N-acceptors on the pyridyl ligands. Hydrogen bonding is one of the core topics of molecular crystal engineering and has been investigated extensively because of its tuneable strength and directional features.102–104,106,108–114 In order to compare the Lewis basicity of the pyridyl ligand in [Fe(η5-C5H4-1-C5H4N)2] towards metal coordination and towards a protic acid, we have also reacted [Fe(η5-C5H4-1-C5H4N)2] with the dicarboxylic organometallic acid [Fe(η5-C5H4COOH)2] obtaining the hydrogen bonded adduct [Fe(η5-C5H4-1-C5H4N)2][Fe(η5-C5H4COOH)2], which will be described later in this Perspective.
Mechanochemical Preparation of Coordination Networks
In this section we will show that coordination polymers with bidentate nitrogen bases can be prepared mechanochemically.126 Nichols, Steed and Raston have explored the use of mechanochemistry in the synthesis of extended supramolecular arrays.127 Grinding of Ni(NO3)2 with 1,10-phenanthroline (phen) resulted in the facile preparation of [Ni(phen)3]2+ accompanied by a dramatic and rapid colour change. Addition of the solid sodium salt of tetrasulfonatocalix[4]arene (tsc) gives two porous π-stacked supramolecular arrays [Ni(phen)3]2[tsc4−]·nH2O and the related [Na(H2O)4(phen)][Ni(phen)3]4 [tsc4−][tsc5−]·nH2O depending on stoichiometry. It has also been reported that the co-grinding of copper(II) acetate hydrate with 1,3-di(4-pyridyl)propane (dpp) gives a gradual colour change from blue to blue–green over ca. 15 min. The resulting material was shown by solid-state NMR spectroscopy to comprise a 1D coordination polymer with water-filled pores. The same host structure, [{Cu(OAc)2}2(µ-dpp)]n, could be obtained from solution containing methanol, acetic acid or ethylene glycol guest species.128We have reported preparation of the coordination polymer Ag[N(CH2CH2)3N]2[CH3COO]·5H2O by co-grinding of silver acetate and [N(CH2CH2)3N] in 1:2 ratio (see Fig. 8). Single crystals suitable for X-ray diffraction were obtained from a water–methanol solution and used to compare calculated and experimental X-ray powder diffractograms. When ZnCl2 is used instead of AgCH3COO in the equimolar reaction with [N(CH2CH2)3N], different products are obtained from solution and solid-state reactions, respectively. The preparation of single crystals of Ag[N(CH2CH2)3N]2[CH3COO]·5H2O was obviously indispensable for the determination of the exact nature of the co-grinding product. In order to do so the powder diffraction pattern computed on the basis of the single-crystal structure was compared with the one measured on the product of the solid-state preparation. Fig. 9 shows that the structure of Zn[N(CH2CH2)3N]Cl2 is based on a one-dimensional coordination network constituted of alternating [N(CH2CH2)3N] and ZnCl2 units, joined by Zn–N bonds. As mentioned above, upon co-grinding of the solid reactants a new Zn compound of unknown stoichiometry is obtained as a powder material. Even though attempts to obtain single crystals of this latter compound have failed, there is a relationship between the compound obtained initially by co-grinding and the one obtained from solution. In fact, the co-grind phase can be partially transformed by prolonged grinding into the known anhydrous phase Zn[N(CH2CH2)3N]Cl2 shown in Fig. 9.
![The coordination network in Ag[N(CH2CH2)3N]2[CH3COO]·5H2O is built up by chains of Ag⋯[N(CH2CH2)3N]⋯Ag⋯[N(CH2CH2)3N]⋯Ag with each silver atom carrying an extra pendant [N(CH2CH2)3N] ligand. An extra coordinated water molecule leads to tetrahedral coordination geometry around the silver centres.](/image/article/2006/DT/b516165g/b516165g-f8.gif) | ||
| Fig. 8 The coordination network in Ag[N(CH2CH2)3N]2[CH3COO]·5H2O is built up by chains of Ag⋯[N(CH2CH2)3N]⋯Ag⋯[N(CH2CH2)3N]⋯Ag with each silver atom carrying an extra pendant [N(CH2CH2)3N] ligand. An extra coordinated water molecule leads to tetrahedral coordination geometry around the silver centres. | ||
![The one-dimensional coordination network present in crystals of Zn[N(CH2CH2)3N]Cl2.](/image/article/2006/DT/b516165g/b516165g-f9.gif) | ||
| Fig. 9 The one-dimensional coordination network present in crystals of Zn[N(CH2CH2)3N]Cl2. | ||
More recently we have applied the same procedure to the preparation of another class of compounds.129 We have explored the solid-state and solution reactions between silver acetate and trans-1,4-diaminocyclohexane [H2NC6H10NH2], dace, a diamine having been little exploited as a divergent ligand in the construction of coordination networks.130–137 The solid-state co-grinding of AgCH3COO and dace in 1 : 1 ratio results in a crystalline powder tentatively formulated as Ag[dace][CH3COO]·xH2O (Scheme 4). Crystallisation of the same compounds from anhydrous MeOH yields two types of products depending on the solvent evaporation conditions: crystals of Ag[dace][CH3COO][MeOH]·0.5H2O, are obtained by crystallisation under argon flow, while slow evaporation in the air results in crystals of Ag[dace][CH3COO]·3H2O. Single-crystal X-ray diffraction experiments have shown that both of these compounds contain two isomeric forms of the coordination network {Ag[dace]+}∞. If the same reaction between AgCH3COO and dace is carried out directly in MeOH–water solution, a third crystalline material is obtained, namely the tetrahydrate Ag[dace][CH3COO]·4H2O. In all cases, correspondence between bulk powder and single crystals was ascertained by comparing computed and observed powder diffractograms.
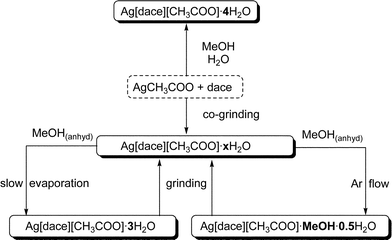 | ||
| Scheme 4 | ||
In terms of chemical composition the three compounds differ only in the degree and nature of solvation. The differences in topology are, however, much more dramatic and the three compounds must be regarded as isomers of the same basic coordination network. The crystal structure of the MeOH·0.5H2O compound is constituted of a two-dimensional coordination network (Fig. 10(a)) formed by the divergent bidentate dace ligand and two silver atoms, which are joined together by an Ag⋯Ag bond of 3.323(1) Å and are asymmetrically bridged by two methanol molecules. There is a close structural relationship between the coordination networks in the MeOH·0.5H2O compound and in the trihydrated compound. This latter structure is built around a zigzag chain Ag(+)⋯[dace]⋯Ag(+)⋯[dace]⋯Ag(+) units as shown in Fig. 10(b). The Ag-atom is coordinated in a linear fashion. A projection perpendicular to the dace planes shows how the zigzag-chains extend in parallel fashion. The Ag(+)⋯[dace]⋯Ag(+)⋯[dace]⋯Ag(+) chains are bridged together via hydrogen bonds involving the N–H donors, the water molecules and the acetate anions. The tetrahydrated species Ag[dace][CH3COO]·4H2O, contains an isomeric form of the coordination networks present in two former compounds. In the trihydrated compound two ligands are in cisoid relative orientation with respect to the silver atom, while in the tetrahydrated compound the two ligands adopt a transoid conformation. This is made possible by the different orientation of the N-atom lone pairs in dace ligand. The acetate anions form a hydrated network and interact with the base and the water molecules.
![(a) Crystal structure of the Ag[dace][CH3COO]·1.5H2O complex where the two-dimensional coordination network is formed by the divergent bidentate dace ligand with silver atoms. (b) Crystal structure of the Ag[dace][CH3COO]·3H2O complex. (c) Crystal structure of the Ag[dace][CH3COO]·4H2O complex where the ligands adopt a transoid relative orientation with respect to the silver atom.](/image/article/2006/DT/b516165g/b516165g-f10.gif) | ||
| Fig. 10 (a) Crystal structure of the Ag[dace][CH3COO]·1.5H2O complex where the two-dimensional coordination network is formed by the divergent bidentate dace ligand with silver atoms. (b) Crystal structure of the Ag[dace][CH3COO]·3H2O complex. (c) Crystal structure of the Ag[dace][CH3COO]·4H2O complex where the ligands adopt a transoid relative orientation with respect to the silver atom. | ||
In summary, different isomers of the same coordination network have been obtained depending on the preparation and crystallisation conditions (Scheme 5). The relationship between supramolecular isomerism and network topology has been discussed.66
 | ||
| Scheme 5 | ||
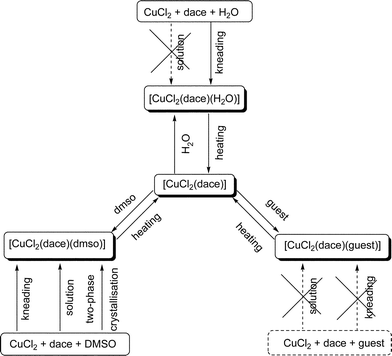
In a further study of mechanochemical utilization of dace we have reported that compound [CuCl2(dace)]∞ can be obtained by thermal treatment of the hydrated compound [CuCl2(dace)(H2O)]∞, which is prepared by kneading of solid CuCl2 and dace in the presence of a small amount of water.138 The structure of [CuCl2(dace)]∞ is not known, since it is insoluble in most organic solvents, which does not permit the growth of single crystals of X-ray quality. However the DMSO adduct [CuCl2(dace)(DMSO)]∞ has been fully characterised by single X-ray diffraction and therefore given some insight of the structure of [CuCl2(dace)]∞. The DMSO adduct can also easily be obtained by kneading solid CuCl2 and dace in the presence of a small amount of DMSO. This compound is formed of 1-D coordination networks, in which the CuCl2 units are bridged by dace ligands in chains (see Fig. 11). Parallel 1-D CuCl2–dace networks form layers and between the layers, the co-crystallised DMSO is intercalated.
![The [CuCl2(dace)]∞ complex forming chains of CuCl2 and dace ligands, where the chains form layers. Between the layers, co-crystallized DMSO is intercalated.](/image/article/2006/DT/b516165g/b516165g-f11.gif) | ||
| Fig. 11 The [CuCl2(dace)]∞ complex forming chains of CuCl2 and dace ligands, where the chains form layers. Between the layers, co-crystallized DMSO is intercalated. | ||
Beside the interesting structural features, compound [CuCl2(dace)(DMSO)]∞ is relevant because of its behaviour upon thermal treatment. When [CuCl2(dace)(DMSO)]∞ is heated to 130 °C it converts to [CuCl2(dace)]∞ as easily ascertained by comparing X-ray diffraction powder diffractograms. From the structure of [CuCl2(dace)(DMSO)]∞ and from the knowledge of its thermal behaviour it is possible to infer that the structure of [CuCl2(dace)]∞ is based on stacking sequence of layers as in [CuCl2(dace)(DMSO)]∞, but “squeezed” at a shorter inter-layer separation as a consequence of DMSO removal. When a guest molecule enters between the layers, the spacing between the CuCl2-dace chains is expanded and the layers are shifted back in position. A series of small molecules can be uptaken/released depending on the preparation method, i.e. kneading, suspension in the liquid guest or kneading followed by suspension. The latter approach is the most productive, when suspended in the desired liquid guest the [CuCl2(dace)]∞ only takes up relatively small molecules (DMSO, acetone, water, methanol, etc.) while by kneading other guest molecules are also taken up. But if [CuCl2(dace)]∞ is first kneaded with a small amount of the desired liquid and then left stirring in the same liquid for 12 h, partial or complete filling of the compound is observed, independently on the guest molecule.
Reversible gas–solid reactions and solid–solid reactions of the zwitterion sandwich complex [CoIII(η5-C5H4COOH)(η5-C5H4COO)]
As mentioned in the Introduction, solid–gas reactions provide another alternative (when not unique) solvent-free route to the preparation of novel materials. In this section we summarize the solvent-free chemistry of the zwitterion sandwich complex [CoIII(η5-C5H4COOH)(η5-C5H4COO)].139 The presence of one –COOH group, which can react with bases, and one –COO(−) group, which can react with acids confers an effective amphoteric behaviour to the complex. As a matter of fact, the molecule undergoes fully reversible gas–solid reactions with the hydrated vapours of a variety of acids (e.g. HCl, CF3COOH, CCl3COOH, CHF2COOH, HBF4, HCOOH and bases (e.g. NH3, NMe3, NH2Me) as well as solid–solid reactions (see Fig. 12) with crystalline alkali salts MX (M = K+, Rb+, Cs+, NH4+; X = Cl−, Br−, I−, PF6−, though not in all permutations of cations and anions).140–145 The zwitterion [CoIII(η5-C5H4COOH)(η5-C5H4COO)] can be quantitatively prepared from the corresponding dicarboxylic cationic acid [CoIII(η5-C5H4COOH)2]+.![The solid–solid reaction and gas–solid reaction of the zwitterion sandwich [CoIII(η5-C5H4COOH)(η5-C5H4COO)] complex with crystalline MX and vapours of difluoroacetic acid, respectively.](/image/article/2006/DT/b516165g/b516165g-f12.gif) | ||
| Fig. 12 The solid–solid reaction and gas–solid reaction of the zwitterion sandwich [CoIII(η5-C5H4COOH)(η5-C5H4COO)] complex with crystalline MX and vapours of difluoroacetic acid, respectively. | ||
Manual grinding of the zwitterion [CoIII(η5-C5H4COOH)(η5-C5H4COO)] with a number of alkali salts MX (M = K+, Rb+, Cs+, NH4+; X = Cl−, Br−, I−, PF6− though not in all permutations of cations and anions, (see below) yields compounds of general formula [CoIII(η5-C5H4COOH)(η5-C5H4COO)]2·M+X−.140,141 Information on the hydrogen-bonding nature and on the relationship between structures in solution and those obtained in the solid state by mechanical grinding were obtained by a combination of solution and solid-state NMR methods. In some cases (M = Rb+, Cs+, X = Cl−, Br−, I−) it was necessary to recur to kneading by adding a few drops of water to the solid mixture in order to obtain the desired product. All compounds of formula [CoIII(η5-C5H4COOH)(η5-C5H4COO)]2·M+X−, (M = K+, Rb+, Cs+, NH4+ X = Br−, I−, PF6−) are isostructural and are characterized by the presence of a supramolecular cage formed by four zwitterionic molecules encapsulating the alkali or ammonium cations. The cage is sustained by O–H⋯O hydrogen bonds between carboxylic –COOH and carboxylate –COO(−) groups, and by C–H⋯O bonds between –CHCp and –CO groups, while the anions are layered in between the cationic complexes, as shown in Fig. 13 in the case of the CsI derivative. It is fascinating to think of the process leading to formation of the cages as a kind of sophisticated solvation based operated by the organometallic complex. The zwitterion is able of “extracting” via O⋯X− interactions the alkali cations from their lattice while the anions are “extruded” and left to interact with the peripheral C–H groups via numerous C–H⋯X interactions. The solid–solid process can thus be seen as the dissolution of one solid (the alkali salt) into a solid solvent.
![A pictorial representation of the process leading from [CoIII(η5-C5H4COOH)(η5-C5H4COO)] and CsI to [CoIII(η5-C5H4COOH)(η5-C5H4COO)]2·Cs+I−.](/image/article/2006/DT/b516165g/b516165g-f13.gif) | ||
| Fig. 13 A pictorial representation of the process leading from [CoIII(η5-C5H4COOH)(η5-C5H4COO)] and CsI to [CoIII(η5-C5H4COOH)(η5-C5H4COO)]2·Cs+I−. | ||
Mechanochemical preparation of hydrogen bonded adducts
Manual grinding of the ferrocenyl dicarboxylic acid complex [Fe(η5-C5H4COOH)2] with nitrogen containing solid bases, namely 1,4-diazabicyclo[2.2.2]octane, 1,4-phenylenediamine, piperazine, trans-1,4-cyclohexanediamine and guanidinium carbonate, generates quantitatively the corresponding organic-organometallic adducts (see Fig. 14(a)).146,147 The case of the adduct [HC6N2H12][Fe(η5-C5H4COOH)(η5-C5H4COO)] (see Fig. 14(b)) is particularly noteworthy because the same product can be obtained in three different ways: (i) by reaction of solid [Fe(η5-C5H4COOH)2] with vapours of 1,4-diazabicyclo[2.2.2]octane (which possesses a small but significant vapour pressure), (ii) by reaction of solid [Fe(η5-C5H4COOH)2] with solid 1,4-diazabicyclo[2.2.2]octane, i.e. by co-grinding of the two crystalline powders, and (iii) by reaction in MeOH solution of the two reactants. Clearly, the fastest process is the solid–solid reaction. It is also interesting to note that the base can be removed by mild treatment regenerating the structure of the starting dicarboxylic acid. The processes imply breaking and reassembling of hydrogen-bonded networks, conformational change from cis to trans of the –COO/–COOH groups on the ferrocene diacid, and proton transfer from acid to base. Crystals suitable for X-ray diffraction were grown via seeding from the solutions of the products originally prepared mechanochemically.![(a) Grinding of the organometallic complex [Fe(η5-C5H4COOH)2] as a solid polycrystalline material with the solid bases 1,4-diazabicyclo[2.2.2]octane, guanidinium carbonate, 1,4-phenylenediamine, piperazine and trans-1,4-cyclohexanediamine generates quantitatively the corresponding adducts [HC6H12N2][Fe(η5-C5H4COOH)(η5-C5H4COO)], [C(NH2)3]2[Fe(η5-C5H4COO)2]·2H2O, [HC6H8N2][Fe(η5-C5H4COOH)(η5-C5H4COO)], [H2C4H10N2][Fe(η5-C5H4COO)2], [H2C6H14N2][Fe(η5-C5H4COO)2]·2H2O. (b) The solid–gas and solid–solid reactions involving 1,4-diazabicyclo[2.2.2]octane with formation of the linear chain.](/image/article/2006/DT/b516165g/b516165g-f14.gif) | ||
| Fig. 14 (a) Grinding of the organometallic complex [Fe(η5-C5H4COOH)2] as a solid polycrystalline material with the solid bases 1,4-diazabicyclo[2.2.2]octane, guanidinium carbonate, 1,4-phenylenediamine, piperazine and trans-1,4-cyclohexanediamine generates quantitatively the corresponding adducts [HC6H12N2][Fe(η5-C5H4COOH)(η5-C5H4COO)], [C(NH2)3]2[Fe(η5-C5H4COO)2]·2H2O, [HC6H8N2][Fe(η5-C5H4COOH)(η5-C5H4COO)], [H2C4H10N2][Fe(η5-C5H4COO)2], [H2C6H14N2][Fe(η5-C5H4COO)2]·2H2O. (b) The solid–gas and solid–solid reactions involving 1,4-diazabicyclo[2.2.2]octane with formation of the linear chain. | ||
In this context it is worth discussing the result of the mechanochemical reaction of the above mentioned complex [Fe(η5–C5H4-1-C5H4N)2] with the dicarboxylic ferrocenyl acid [Fe(η5-C5H4COOH)2]. The supramolecular structure of the hydrogen bond adduct [Fe(η5-C5H4-1-C5H4N)2][Fe(η5-C5H4COOH)2] is shown in Fig. 15. It can be seen that [Fe(η5-C5H4-1-C5H4N)2] and [Fe(η5-C5H4COOH)2] establishes a twin hydrogen-bonding interaction forming a sort of ferrocenyl dimer. The O⋯N separations [O(2)⋯N(1) 2.593(5), O(4)⋯N(2) 2.569(5) Å] are in agreement with the presence of conventional hydrogen bonds. It is interesting to observe that, judging from the diffraction data, no proton transfer from the –COOH groups to the N-sites takes place. Hence the two hydrogen-bonding interactions ought to be described as neutral O–H⋯N rather than as “charge assisted” (−)O⋯H–N(+)hydrogen bonds.107,148–152 Both the two COOH groups in the diacid and the pyridine groups in [Fe(η5-C5H4-1-C5H4N)2] are eclipsed. The arrangement is thus topologically related to that observed for the class of “complexes of complexes” discussed above with the exception of the (ZnIIchloride)24+ derivative. It is interesting to note that the preference for an eclipsed conformation of the pyridyl ligands is maintained in the formation of the hydrogen bond adduct. In principle, [Fe(η5-C5H4-1-C5H4N)2][Fe(η5-C5H4COOH)2] could possess an alternative network structure in the solid state based on 1-D chains of alternating pyridylcarboxylic ligands. This arrangement is fairly common when ferrocenyl dicarboxylic acid is used. It has been observed in hydrogen bonded adducts with bis-amidines153 and with other bis-amines, such as 1,4-diazabicyclo[2.2.2]octane, C6H12N2, DABCO.146,154 In all these cases, however, the geometry of the base did not allow cyclization and forced the system to chain formation, while the conformational freedom of [Fe(η5-C5H4-1-C5H4N)2] permits, in principle, both linear chains and rings.
![The solid-state structure of [Fe(η5-C5H4-1-C5H4N)2][Fe(η5-C5H4COOH)2] showing the twin hydrogen-bonding interaction which form the ferrocenyl dimer.](/image/article/2006/DT/b516165g/b516165g-f15.gif) | ||
| Fig. 15 The solid-state structure of [Fe(η5-C5H4-1-C5H4N)2][Fe(η5-C5H4COOH)2] showing the twin hydrogen-bonding interaction which form the ferrocenyl dimer. | ||
A similar situation is seen with the diacid [Fe(η5-C5H4COOH)2] itself.155–163 Contrary to most dicarboxylic acids that form chains linked by carboxylic rings, the ferrocenyl diacid forms cyclic dimers joined by a twin carboxylic ring in both its known polymorphic modifications (monoclinic and triclinic). It is noteworthy that the analogy between [Fe(η5-C5H4-1-C5H4N)2][Fe(η5-C5H4COOH)2] and [Fe(η5-C5H4COOH)2] is not confined to the dimeric structure: the supramolecular arrangement in the solid state is also extremely similar. The effect of mechanical mixing of solid dicarboxylic acids HOOC(CH2)nCOOH (n = 1–7) of variable chain length together with the solid base 1,4-diazabicyclo[2.2.2]octane, C6H12N2, to generate the corresponding salts or cocrystals of formula [N(CH2CH2)3N]–H–[OOC(CH2)nCOOH] (n = 1–7) has also been investigated.164
Mechanochemical preparation of supramolecular crown ether adducts
In a further extension of the exploratory work in the use of mechanochemical methods to prepare hydrogen bonded supramolecular adducts we have used crown ethers to capture alkali metal cations and the ammonium cation in extended hydrogen bonded networks.165 Crown ethers complexes have been the subject of an enormous number of studies because of the interest in ion recognition, complexation and in self-assembly processes.166–170 In the study ammonium hydrate sulfate salts were used since the presence of hydrogen bonds between ions is a relevant supramolecular issue and hydrogen sulfate salts have found applications in a number of devises such as H2 and H2O sensors, fuel and steam cells and high energy density batteries.171–174 Manual co-grinding of solid 18-crown[6] and solid [NH4][HSO4] in the air leads to formation of the crown ether complex 18-crown[6]·[NH4][HSO4]·2H2O (Fig. 16), the water molecules being taken up from ambient humidity during grinding. The complex has been fully structurally characterised by single-crystal X-ray determination. In the complex the ammonium cation is trapped via Ocrown⋯H–N hydrogen bonds by the crown ethers, while on the exposed side it interacts with the hydrogen sulfate anion. The sulfate anion and the water molecules also interacts via hydrogen bonding forming a ribbon that is sandwiched between 18-crown[6]·[NH4]+ units. Hydrogen bonds are also observed between water molecules and oxygen atoms in the crown ether.![The solid-state structure of the crown ether complex 18-crown[6]·[NH4][HSO4]·2H2O.](/image/article/2006/DT/b516165g/b516165g-f16.gif) | ||
| Fig. 16 The solid-state structure of the crown ether complex 18-crown[6]·[NH4][HSO4]·2H2O. | ||
The crown ether 15-crown[5] is a liquid in room temperature, so when it is kneaded instead of grinded, with the ammonium hydrate sulfate, a similar reaction as for the 18-crown[6] takes place. The product, (15-crown[5])3·[NH4]2[HSO4]3·H2O, also fully structurally determined by single-crystal X-ray determination, is also obtained when the reaction takes place in solution. The (15-crown[5])3·[NH4]2[HSO4]3·H2O is reminiscent of that of the 18-crown[6] because of the formation of hydrogen bonded ribbons intercalated between the crown ether layers. The difference between the two adducts are, however, the two different types of interactions between the ammonium cation and the crown ether that is presence in the 15-crown[5] adduct. One ammonium cation is sandwiched between two crown ether units, while the other is linked to the hydrogen sulfate anion by N–H⋯O hydrogen bonds. In the 15-crown[5] adduct also a hydrogen bonded [H3O]+ ion is needed to neutralise the overall charge.
Conclusions and outlook
Crystal engineering amounts to the deliberate construction of a molecular solid1 (whether a molecular complex, an adduct or a co-crystal)175,176 that can perform desired functions, hence it is conceptually related to the construction of a supermolecule.166,177 In both molecular crystals and supermolecules the collective properties depend on the aggregation via intermolecular bonds of two or more component units. These supramolecular interactions can be coordination bonds between ligands and metal centres and non-covalent bonds between neutral molecules or ions or, of course, any of their combinations. Processes that lead to such non-covalent bonds from reactants to products, either via breaking or forming of intermolecular bonds, are therefore supramolecular reactions. In this Perspective article we have shown that non-covalent bonds can be broken and formed in a controlled way by reactions that do not imply the use of solvent but that can be carried out directly between two crystalline solids or between a crystalline solid and a vapour. Reactions of this type have been the subject of investigation for decades in the fields of organic and of inorganic chemistry. Although solid–gas and solid–solid reactions are at the basis of a number of industrial processes that range from preparation of pharmaceutical compounds178,179 to inorganic alloying,24 they still enjoy little popularity in the field or organometallic and coordination chemistry.180,181 This is probably due, on the one hand, to the fact that crystals are depicted (even at the level of crystallography courses) as rigid, stiff, fragile materials that are good for little else beside structural analysis, and, on the other hand, to the belief that molecular crystals, being held together by non-covalent interactions, cannot compete with covalent or ionic inorganic solids in terms of cohesion and stability and are not the best materials for gas uptake and/or mechanical treatment.Our experience is that adequately chosen crystalline materials can withstand reversible gas–solid reactions with vapours of both acidic and basic substances as well as mechanically activated reactions with other molecular crystals and inorganic salts. Primarily, the interest in solvent-free conditions stems from the possibility of obtaining the same product as that from solution without solvent because the process is cheaper, less time consuming and often more environmentally friendly. On the other hand one may be interested in solvent-free conditions for the possibility of obtaining products not otherwise accessible from solvents. In this latter case, however, one is often faced with the problem of characterization, because the lack of single crystals complicates the matter significantly and ab initio structural determination from powder diffraction data alone is not yet a valid alternative with complex structures such as those described above.
In this Perspective, we have confined ourselves to essentially four classes of reactions involving organometallic molecular crystals or coordination compounds as reactants: (i) reactions between a hydrogen bonded molecular crystal and a vapour with formation of hydrogen bonded supramolecular adducts, (ii) reactions leading to formation of covalent bonds for the preparation of building blocks, (iii) reactions between hydrogen bonded molecular crystals to produce new molecular crystals based on hydrogen bonds, and (iv) reactions between a molecular crystals and ionic crystals leading to “solid-state solvation”. Clearly, all these reactions (perhaps with the exception of those of type (ii) are diffusion controlled and are not necessarily reactions in the solid state as mechanical stress may cause local melting, co-grinding may form an intermediate eutectic phase, and kneading probably generates locally hypersaturated solutions wherefrom crystals of the new phase nucleate. In all these cases the crystal lattice is destroyed and reformed through recrystallization. In such processes hydrogen bonds, π-stacking, van der Waals, ion pairing interactions etc. in are broken and formed through the reaction process leading to formation of supramolecular compounds or hybrid molecular crystals.
On closing, it may be useful to stress that reactions involving solid coordination and organometallic compounds represent “green chemistry” ways to the preparation of metal containing materials since recovery, storage and disposal of solvents are not required.182 Furthermore solvent-free reactions often lead to very pure products and reduce the formation of solvate species and may thus be exploited in the quest for elusive crystal polymorphs.183–189 These might be useful notions for crystal engineers and solid-state chemists.
Acknowledgements
We thank MIUR (PRIN2004 and FIRB2001) the University of Bologna, and Bertil Lundqvist Foundation (A. P.) for financial support.References
- G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed.
- D. Braga, F. Grepioni and G. R. Desiraju, Chem. Rev., 1998, 98, 1375–1405 CrossRef CAS.
- J. Chem. Soc., Dalton Trans., 2000, 3705–3998, Proceedings of the Dalton Discussion on Inorganic Crystal Engineering Search PubMed.
- M. D. Hollingsworth, Science, 2002, 295, 2410–2413 CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- D. Braga, F. Grepioni and A. G. Orpen, Crystal Engineering: from molecules and crystals to materials, Kluwer Academic Publishers, Dordrecht, 1999 Search PubMed.
- D. Braga, Chem. Commun., 2003, 2751–2754 RSC.
- L. Addadi and M. Geva, CrystEngComm, 2003, 5, 140–146 RSC.
- J. Aizenberg, G. Lambert, S. Weiner and L. Addadi, J. Am. Chem. Soc., 2002, 124, 32–39 CrossRef CAS.
- E. Beniash, J. Aizenberg, L. Addadi and S. Weiner, Proc. R. Soc. London, Ser. B, 1997, 264, 461–465 CrossRef CAS.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959–970 CrossRef CAS.
- J. Johnston, H. E. Merwin and E. D. Williamson, Am. J. Sci., 1916, 41, 473–512 Search PubMed.
- S. Raz, S. Weiner and L. Addadi, Adv. Mater., 2000, 12, 38–42 CrossRef CAS.
- E. Loste and F. C. Meldrum, Chem. Commun., 2001, 901–902 RSC.
- J. Aizenberg, G. Lambert, L. Addadi and S. Weiner, Adv. Mater., 1996, 8, 222–226 CAS.
- D. Braga, Angew. Chem., Int. Ed., 2003, 42, 5544–5546 CrossRef CAS.
- D. Braga and F. Grepioni, Chem. Soc. Rev., 2000, 29, 229–238 RSC.
- D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002–4011 CrossRef CAS.
- D. Braga, D. D'Addario, L. Giaffreda Stefano, L. Maini, M. Polito and F. Grepioni, Top. Curr. Chem., 2005, 254, 71–94 CAS.
- D. Braga and F. Grepioni, Chem. Commun., 2005, 3635–3645 RSC.
- D. Braga and F. Grepioni, J. Chem. Soc., Dalton Trans., 1999, 1–8 RSC.
- D. Braga and F. Grepioni, Coord. Chem. Rev., 1999, 183, 19–41 CrossRef CAS.
- D. Braga, L. Maini, M. Polito, L. Scaccianoce, G. Cojazzi and F. Grepioni, Coord. Chem. Rev., 2001, 216–217, 225–248 CrossRef.
- V. V. Boldyrev and K. Tkacova, J. Mater. Synth. Process., 2000, 8, 121–132 CrossRef CAS.
- J. F. Fernandez-Bertran, Pure Appl. Chem., 1999, 71, 581–586 CrossRef CAS.
- G. Kaupp, Compr. Supramol. Chem., 1996, 8, 381–423 Search PubMed.
- G. Kaupp, CrystEngComm, 2003, 5, 117–133 RSC.
- J. M. Cairney, S. G. Harris, L. W. Ma, P. R. Munroe and E. D. Doyle, J. Mater. Sci., 2004, 39, 3569–3575 CrossRef CAS.
- G. Bettinetti, M. R. Caira, A. Callegari, M. Merli, M. Sorrenti and C. Tadini, J. Pharm. Sci., 2000, 89, 478–489 CrossRef CAS.
- Y. A. Kim, T. Hayashi, Y. Fukai, M. Endo, T. Yanagisawa and M. S. Dresselhaus, Chem. Phys. Lett., 2002, 355, 279–284 CrossRef CAS.
- K. Wieczorek-Ciurowa, K. Gamrat and K. Fela, Solid State Ionics, 2003, 164, 193–198 CrossRef CAS.
- N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372–2373 RSC.
- S. Watano, T. Okamoto, M. Tsuhari, I. Koizumi and Y. Osako, Chem. Pharm. Bull., 2002, 50, 341–345 CrossRef CAS.
- S. Watano, J. Furukawa, K. Miyanami and Y. Osako, Adv. Powder Technol., 2001, 12, 427–441 Search PubMed.
- N. Morin, A. Chilouet, J. Millet and J. C. Rouland, J. Therm. Anal. Calorim., 2000, 62, 187–201 CrossRef CAS.
- G. Bruni, A. Marini, V. Berbenni, R. Riccardi and M. Villa, J. Inclusion Phenom. Macrocyclic Chem., 1999, 35, 517–530 Search PubMed.
- F. Taneri, T. Gueneri, Z. Aigner and M. Kata, J. Inclusion Phenom. Macrocyclic Chem., 2002, 44, 257–260 Search PubMed.
- R. Saikosin, T. Limpaseni and P. Pongsawasdi, J. Inclusion Phenom. Macrocyclic Chem., 2002, 44, 191–196 Search PubMed.
- T. L. Threlfall, Analyst, 1995, 120, 2435–2460 RSC.
- N. Kubota, N. Doki, M. Yokota and D. Jagadesh, J. Chem. Eng. Jpn., 2002, 35, 1063–1071 Search PubMed.
- D. Braga and F. Grepioni, in Crystal Design, ed. G. R. Desiraju, John Wiley & Sons, Weinheim, 2003 Search PubMed.
- W. C. McCrone, in Polymorphism in Physics and Chemistry of the Organic Solid State, ed. D. Fox, M. M. Labes and A. Weissemberg, Interscience, New York, 1965, vol. 2, p. 726 Search PubMed.
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, Oxford, 2002 Search PubMed.
- P. Seiler and J. D. Dunitz, Acta Crystallogr., Sect. B, 1982, 38, 1741–1745 CrossRef.
- D. Braga, G. Cojazzi, D. Paolucci and F. Grepioni, CrystEngComm, 2001, 3, 159–161 RSC.
- R. J. Davey, N. Blagden, G. D. Potts and R. Docherty, J. Am. Chem. Soc., 1997, 119, 1767–1772 CrossRef CAS.
- H. Koshima and M. Miyauchi, Cryst. Growth Des., 2001, 1, 355–357 CrossRef CAS.
- J. D. Dunitz and J. Bernstein, Acc. Chem. Res., 1995, 28, 193–200 CrossRef CAS.
- V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, Chem. Commun., 2002, 1606–1607 RSC.
- M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645–5647 CrossRef CAS.
- M. Fujita, J. Yazaki and K. Ogura, Chem. Lett., 1991, 1031–1032 CAS.
- A. Orita, L. Jiang, T. Nakano, N. Ma and J. Otera, Chem. Commun., 2002, 1362–1363 RSC.
- C. R. Woods, M. Benaglia, F. Cozzi and J. S. Siegel, Angew. Chem., Int. Ed. Engl., 1996, 35, 1830–1833 CrossRef CAS.
- R. Annunziata, M. Benaglia, M. Cinquini, F. Cozzi, C. R. Woods and J. S. Siegel, Eur. J. Org. Chem., 2001, 173–180 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- T. Ishiyama, H. Kizaki, T. Hayashi, A. Suzuki and N. Miyaura, J. Org. Chem., 1998, 63, 4726–4731 CrossRef CAS.
- D. Villemin and F. Caillot, Tetrahedron Lett., 2001, 42, 639–642 CrossRef CAS.
- G. W. Kabalka, R. M. Pagni and C. M. Hair, Org. Lett., 1999, 1, 1423–1425 CrossRef CAS.
- M. Melucci, G. Barbarella and G. Sotgiu, J. Org. Chem., 2002, 67, 8877–8884 CrossRef CAS.
- D. Braga, D. D'Addario, M. Polito and F. Grepioni, Organometallics, 2004, 23, 2810–2812 CrossRef CAS.
- D. Braga, M. Polito, M. Bracaccini, D. D'Addario, E. Tagliavini, L. Sturba and F. Grepioni, Organometallics, 2003, 22, 2142–2150 CrossRef CAS.
- D. Braga and F. Grepioni, Acc. Chem. Res., 2000, 33, 601–608 CrossRef CAS.
- A. M. Garcia, D. M. Bassani, J.-M. Lehn, G. Baum and D. Fenske, Chem.–Eur. J., 1999, 5, 1234–1238 CrossRef CAS.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS.
- S. R. Batten, CrystEngComm, 2001, 18, 1–17 RSC.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS.
- A. J. Blake, N. R. Champness, P. Hubberstey, W.-S. Li, M. A. Withersby and M. Schroder, Coord. Chem. Rev., 1999, 183, 117–138 CrossRef CAS.
- S. R. Batten, B. F. Hoskins and R. Robson, Chem.–Eur. J., 2000, 6, 156–161 CrossRef CAS.
- J. Lu, B. Moulton, M. J. Zaworotko and S. A. Bourne, Chem. Commun., 2001, 861–862 RSC.
- B. Rather and M. J. Zaworotko, Chem. Commun., 2003, 830–831 RSC.
- B. Moulton, H. Abourahma, M. W. Bradner, J. Lu, G. J. McManus and M. J. Zaworotko, Chem. Commun., 2003, 1342–1343 RSC.
- M. Fujita, Chem. Soc. Rev., 1998, 27, 417–425 RSC.
- B. Olenyuk, A. Fechtenkotter and P. J. Stang, J. Chem. Soc., Dalton Trans., 1998, 1707–1728 RSC.
- M. D. Ward, Science, 2003, 300, 1104–1105 CrossRef CAS.
- D. Braga, G. R. Desiraju, J. S. Miller, A. G. Orpen and S. L. Price, CrystEngComm, 2002, 4, 500–509 RSC.
- G. Ferey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guegan, Chem. Commun., 2003, 2976–2977 RSC.
- F. A. Cotton, C. Lin and C. A. Murillo, J. Chem. Soc., Dalton Trans., 2001, 499–501 RSC.
- F. A. Cotton, C. Lin and C. A. Murillo, Chem. Commun., 2001, 11–12 RSC.
- W. Mori and S. Takamizawa, J. Solid State Chem., 2000, 152, 120–129 CrossRef CAS.
- L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, CrystEngComm, 2002, 4, 121–129 RSC.
- Inorganic Materials, ed. D. W. Bruce and D. O'Hare, Wiley, Chichester, 1992 Search PubMed.
- S. R. Marder, Inorg. Mater., 1992, 115–164 CAS.
- N. J. Long, Angew. Chem., Int. Ed. Engl., 1995, 34, 21–38 CrossRef CAS.
- T. J. Marks and M. A. Ratner, Angew. Chem., Int. Ed. Engl., 1995, 34, 155–173 CrossRef CAS.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev. (Washington, DC), 1994, 94, 195–242 Search PubMed.
- O. Khan, Molecular Magnetism, VCH, New York, 1993 Search PubMed.
- D. Gatteschi, Adv. Mater., 1994, 6, 635–645 CAS.
- J. S. Miller and A. J. Epstein, Chem. Eng. News, 1995, 73, 30–41 CAS.
- J. S. Miller, Angew. Chem., Int. Ed., 2003, 42, 27–29 CrossRef CAS.
- L. Pan, M. B. Sander, X. Huang, J. Li, M. Smith, E. Bittner, B. Bockrath and J. K. Johnson, J. Am. Chem. Soc., 2004, 126, 1308–1309 CrossRef CAS.
- D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1–19 RSC.
- H. Plenio and D. Burth, Organometallics, 1996, 15, 4054–4062 CrossRef CAS.
- E. C. Constable, A. J. Edwards, M. D. Marcos, P. R. Raithby, R. Martinez-Manez and M. J. L. Tendero, Inorg. Chim. Acta, 1994, 224, 11–14 CrossRef CAS.
- C. D. Hall, T. K. U. Truong and J. H. R. Tucker, Chem. Commun., 1997, 2195–2196 RSC.
- C. B. Aakeroy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- K. Biradha, CrystEngComm, 2003, 5, 374–384 RSC.
- H. W. Roesky and M. Andruh, Coord. Chem. Rev., 2003, 236, 91–119 CrossRef CAS.
- A. M. Beatty, Coord. Chem. Rev., 2003, 246, 131–143 CrossRef CAS.
- E. A. Bruton, L. Brammer, F. C. Pigge, C. B. Aakeroey and D. S. Leinen, New J. Chem., 2003, 27, 1084–1094 RSC.
- D. Braga, M. Polito, D. D'Addario, E. Tagliavini, D. M. Proserpio, F. Grepioni and J. W. Steed, Organometallics, 2003, 22, 4532–4538 CrossRef CAS.
- R. Schneider, M. W. Hosseini, J.-M. Planeix, A. De Cian and J. Fischer, Chem. Commun., 1998, 1625–1626 RSC.
- G. Desiraju and T. Steiner, The Weak Hydrogen Bond: Applications to Structural Chemistry and Biology, Oxford Univ. Press, New York, 1999 Search PubMed.
- D. Braga, F. Grepioni, K. Biradha, V. R. Pedireddi and G. R. Desiraju, J. Am. Chem. Soc., 1995, 117, 3156–3166 CrossRef CAS.
- M. J. Zaworotko, Chem. Soc. Rev., 1994, 23, 283–288 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- M. J. Calhorda, Chem. Commun., 2000, 801–809 RSC.
- D. Braga, L. Maini, M. Polito and F. Grepioni, Struct. Bonding (Berlin), 2004, 111, 1–32 CAS.
- C. Janiak, Angew. Chem., Int. Ed. Engl., 1997, 36, 1431–1434 CrossRef CAS.
- S. George, A. Nangia, M. Bagieu-Beucher, R. Masse and J.-F. Nicoud, New J. Chem., 2003, 27, 568–576 RSC.
- M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC.
- J. Hulliger, Chem.–Eur. J., 2002, 8, 4578–4586 CrossRef CAS.
- L. Brammer, M. D. Burgard, M. D. Eddleston, C. S. Rodger, N. P. Rath and H. Adams, CrystEngComm, 2002, 4, 239–248 RSC.
- L. Brammer, Dalton Trans., 2003, 3145–3157 RSC.
- A. D. Burrows, Struct. Bonding (Berlin), 2004, 108, 55–96 CAS.
- J. Organomet. Chem., 2001, 637–639, 1–875 Search PubMed (whole issue).
- M. W. Hosseini and A. De Cian, Chem. Commun., 1998, 727–733 RSC.
- D. Braga and F. Grepioni, Chem. Commun., 1996, 571–578 RSC.
- T.-Y. Liu, Y. J. Chen, C.-C. Tai and K. S. Kwan, Inorg. Chem., 1999, 38, 674–679 CrossRef CAS.
- K. Hutchison, J. C. Morris, T. A. Nile, J. L. Walsh, D. W. Thompson, J. D. Petersen and J. R. Schoonover, Inorg. Chem., 1999, 38, 2516–2523 CrossRef CAS.
- E. M. Barranco, O. Crespo, M. C. Gimeno, P. G. Jones, A. Laguna and M. D. Villacampa, J. Organomet. Chem., 1999, 592, 258–264 CrossRef CAS.
- E. C. Constable, A. J. Edwards, R. Martinez-Manez, P. R. Raithby and A. M. W. C. Thompson, J. Chem. Soc., Dalton Trans., 1994, 645–650 RSC.
- L. Brammer, M. D. Burgard, C. S. Rodger, J. K. Swearingen and N. P. Rath, Chem. Commun., 2001, 2468–2469 RSC.
- D. Braga, M. Polito, M. Bracaccini, D. D'Addario, E. Tagliavini, D. M. Proserpio and F. Grepioni, Chem. Commun., 2002, 1080–1081 RSC.
- C.-Y. Su, B.-S. Kang, Q.-G. Wang and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 2000, 1831–1833 RSC.
- G. W. Eastland, M. A. Mazid, D. R. Russell and M. C. R. Symons, J. Chem. Soc., Dalton Trans., 1980, 1682–1687 RSC.
- D. Braga, S. L. Giaffreda, F. Grepioni and M. Polito, CrystEngComm, 2004, 6, 458–462 Search PubMed.
- P. J. Nichols, C. L. Raston and J. W. Steed, Chem. Commun., 2001, 1062–1063 RSC.
- W. J. Belcher, C. A. Longstaff, M. R. Neckenig and J. W. Steed, Chem. Commun., 2002, 1602–1603 RSC.
- D. Braga, M. Curzi, F. Grepioni and M. Polito, Chem. Commun., 2005, 2915–2917 RSC.
- J. L. Stark, A. L. Rheingold and E. A. Maatta, J. Chem. Soc., Chem. Commun., 1995, 1165–1166 RSC.
- N. L. Rosi, M. Eddaoudi, J. Kim, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2002, 41, 284–287 CrossRef.
- O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and M. Yaghi Omar, Science, 2002, 295, 469–472 CrossRef.
- N. L. Rosi, M. Eddaoudi, J. Kim, M. O'Keeffe and O. M. Yaghi, CrystEngComm, 2002, 4, 401–404 RSC.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1130 CrossRef CAS.
- M. E. Braun, C. D. Steffek, J. Kim, P. G. Rasmussen and O. M. Yaghi, Chem. Commun., 2001, 2532–2533 RSC.
- D. Braga, M. Curzi, A. Johansson, M. Polito, K. Rubini and F. Grepioni, Angew. Chem., Int. Ed., 2006, 45, 142–146 CrossRef CAS.
- D. Braga, L. Maini, M. Polito and F. Grepioni, Organometallics, 1999, 18, 2577–2579 CrossRef CAS.
- D. Braga, G. Cojazzi, D. Emiliani, L. Maini and F. Grepioni, Chem. Commun., 2001, 2272–2273 RSC.
- D. Braga, G. Cojazzi, D. Emiliani, L. Maini and F. Grepioni, Organometallics, 2002, 21, 1315–1318 CrossRef CAS.
- D. Braga, L. Maini, M. Mazzotti, K. Rubini and F. Grepioni, CrystEngComm, 2003, 5, 154–158 RSC.
- D. Braga, L. Maini, M. Mazzotti, K. Rubini, A. Masic, R. Gobetto and F. Grepioni, Chem. Commun., 2002, 2296–2297 RSC.
- G. Kaupp, M. R. Naimi-Jamal, L. Maini, F. Grepioni and D. Braga, CrystEngComm, 2003, 5, 474–479 RSC.
- D. Braga, G. Cojazzi, D. Emiliani, L. Maini, M. Polito, R. Gobetto and F. Grepioni, CrystEngComm, 2002, 4, 277–281 RSC.
- D. Braga, L. Maini, M. Polito, L. Mirolo and F. Grepioni, Chem. Commun., 2002, 2960–2961 RSC.
- D. Braga, L. Maini, M. Polito, L. Mirolo and F. Grepioni, Chem.–Eur. J., 2003, 9, 4362–4370 CrossRef CAS.
- C. C. Wilson, Acta Crystallogr., Sect. B, 2001, 57, 435–439 CrossRef CAS.
- T. Steiner, I. Majerz and C. C. Wilson, Angew. Chem., Int. Ed., 2001, 40, 2651–2654 CrossRef CAS.
- D. Wiechert and D. Mootz, Angew. Chem., Int. Ed., 1999, 38, 1974–1976 CrossRef CAS.
- D. Braga, L. Maini, S. L. Giaffreda, F. Grepioni, M. R. Chierotti and R. Gobetto, Chem.–Eur. J., 2004, 10, 3261–3269 CrossRef CAS.
- D. Braga, K. Rubini and L. Maini, CrystEngComm, 2004, 6, 236–238 RSC.
- D. Braga, L. Maini, F. Grepioni, A. De Cian, O. Felix, J. Fischer and M. Wais Hosseini, New J. Chem., 2000, 24, 547–553 RSC.
- C. M. Zakaria, G. Ferguson, A. J. Lough and C. Glidewell, Acta Crystallogr., Sect. B, 2002, 58, 786–802 CrossRef.
- F. Takusagawa and T. F. Koetzle, Acta Crystallogr., Sect. B, 1979, 35, 2888–2896 CrossRef.
- G. J. Palenik, Inorg. Chem., 1969, 8, 2744–2749 CrossRef CAS.
- S. Takamizawa, E.-i. Nakata, T. Saito, T. Akatsuka and K. Kojima, CrystEngComm, 2004, 6, 197–199 RSC.
- S. Takamizawa, E.-i. Nakata and T. Saito, Angew. Chem., Int. Ed., 2004, 43, 1368–1371 CrossRef CAS.
- S. Takamizawa, E.-i. Nakata and T. Saito, CrystEngComm, 2004, 6, 39–41 RSC.
- S. Takamizawa, E.-i. Nakata, H. Yokoyama, K. Mochizuki and W. Mori, Angew. Chem., Int. Ed., 2003, 42, 4331–4334 CrossRef CAS.
- E. Le Fur, E. Demers, T. Maris and J. D. Wuest, Chem. Commun., 2003, 2966–2967 RSC.
- D. Laliberte, T. Maris and J. D. Wuest, J. Org. Chem., 2004, 69, 1776–1787 CrossRef CAS.
- O. Saied, T. Maris and J. D. Wuest, J. Am. Chem. Soc., 2003, 125, 14956–14957 CrossRef CAS.
- D. Braga, L. Maini, G. de Sanctis, K. Rubini, F. Grepioni, M. R. Chierotti and R. Gobetto, Chem.–Eur. J., 2003, 9, 5538–5548 CrossRef CAS.
- D. Braga, M. Curzi, M. Lusi and F. Grepioni, CrystEngComm, 2005, 7, 276–278 RSC.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry: A Concise Introduction, John Wiley & Sons, Ltd., London, 2000 Search PubMed.
- M. Calleja, S. A. Mason, P. D. Prince, J. W. Steed and C. Wilkinson, New J. Chem., 2003, 27, 28–31 RSC.
- P. C. Junk, B. J. McCool, B. Moubaraki, K. S. Murray, L. Spiccia, J. D. Cashion and J. W. Steed, J. Chem. Soc., Dalton Trans., 2002, 1024–1029 RSC.
- J. L. Atwood, K. T. Holman and J. W. Steed, Chem. Commun., 1996, 1401–1407 RSC.
- J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS.
- J. Lipkowski, B. Baranowski and A. Lunden, Pol. J. Chem., 1993, 67, 1867–1876 CAS.
- S. M. Haile, Mater. Res. Soc. Symp. Proc., 1999, 547, 315–326 CAS.
- S. M. Haile, D. A. Boysen, C. R. I. Chisholm and R. B. Merle, Nature, 2001, 410, 910–913 CrossRef CAS.
- C. R. I. Chisholm and S. M. Haile, Solid State Ionics, 2000, 136–137, 229–241 CrossRef CAS.
- G. R. Desiraju, CrystEngComm, 2003, 5, 466–467 RSC.
- J. D. Dunitz, CrystEngComm, 2003, 5, 506 RSC.
- J. M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed.
- A. Burger, in Topics in Pharmaceutical Science, ed. D. Breimer and P. Speiser, Elsevier, Amsterdam, 1983, p. 347 Search PubMed.
- S. R. Byrn, Solid-State Chemistry of Drugs, Academic Press, New York, 1982 Search PubMed.
- M. Albrecht, M. Lutz, A. L. Spek and G. van Koten, Nature, 2000, 406, 970–974 CrossRef CAS.
- M. Albrecht, R. A. Gossage, M. Lutz, A. L. Spek and G. van Koten, Chem.–Eur. J., 2000, 6, 1431–1445 CrossRef CAS.
- P. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS.
- K. Tanaka and F. Toda, Solvent-Free Organic Synthesis, Wiley-VCH, London, 2003 Search PubMed.
- D. Bradley, Chem. Br., 2002, 38, 42–45 Search PubMed.
- G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 2159–2169 RSC.
- G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701–8708 CrossRef CAS.
- F. Toda, CrystEngComm, 2002, 4, 215–222 RSC.
- L. R. Nassimbeni, Acc. Chem. Res., 2003, 36, 631–637 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |