Development of a convenient new synthetic route to [3]ferrocenophanones
Ludger
Tebben
a,
Markus
Neumann
a,
Gerald
Kehr
a,
Roland
Fröhlich†
a,
Gerhard
Erker
*a,
Serena
Losi‡
b and
Piero
Zanello‡
b
aOrganisch-Chemisches Institut der Universität Münster, Corrensstr. 40, D-48149, Münster, Germany. E-mail: erker@uni-muenster.de; Fax: +49 251-83 36503
bUniversitá di Siena, Dipartímento di Chimica, Via Aldo Moro, I-53100, Siena, Italy. E-mail: zanello@unisi.it; Fax: + 39 0577-234254
First published on 27th January 2006
Abstract
[3]Ferrocenophanone rac-8 was prepared by several non-Friedel–Crafts pathways starting from a Mannich-type coupling of 1,1′-diacetylferrocene followed by catalytic hydrogenation. Hydride abstraction from the resulting α-dimethylamino[3]ferrocenophane rac-14 with B(C6F5)3 followed by hydrolysis gave the ketone rac-8. Several variants of the Sommelet reaction, using ethylglyoxylate, formaldehyde or hexamethylenetetramine (urotropine) as the “oxidizing” reagent gave the α-[3]ferrocenophanone 8 in good to excellent yield. Some variants of these reactions were also used for the preparation of the pure enantiomer (R)-8. The electrochemical behaviour of 8 has been investigated and compared with related derivatives.
Introduction
The [3]ferrocenophanones are important starting materials for a great variety of [3]ferrocenophane systems, including a number of ligand systems for use e.g. in asymmetric catalysis.1,2 The parent β-isomer (2) (Scheme 1) is probably best available by means of an intramolecular Dieckmann condensation sequence3 whereas the unsubstituted α-[3]ferrocenophanone isomer (4) has commonly been prepared by a Friedel–Crafts reaction. Treatment of ferrocenylpropionic acid (3) with trifluoroacetic anhydride leads to ring closure with formation of the three-carbon bridge between the pair of Cp-rings at the ferrocene nucleus to yield 4.4,5 This sequence was also used for the construction of multibridged [3]nferrocenophane systems (n = 2 or 3).6,7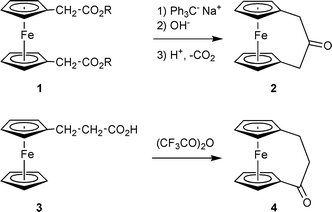 | ||
| Scheme 1 | ||
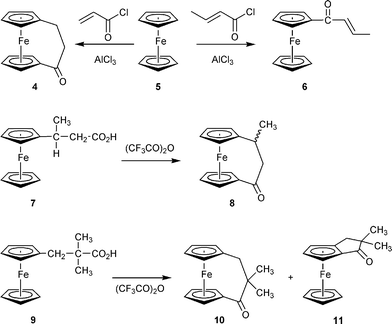
The formation of substituted derivatives of 4 is sometimes more difficult to achieve by the Friedel–Crafts routes. Here are some typical examples. Whereas treatment of ferrocene (5) with acrylic acid chloride/AlCl3 cleanly leads to the formation of 4,8 the corresponding reaction of 5 with crotonoyl chloride only gives the open product 6 (Scheme 2). This does not ring close under subsequent treatment with aluminium chloride, even under forcing conditions. The corresponding reaction between ferrocene and methacryloyl chloride gave mixtures of [3]ferrocenophanone and open products. 3-Ferrocenylbutanoic acid 7 could be converted to the methylated [3]ferrocenophanone 8,9 whereas the sterically more hindered derivative 9 gave a ca. 1 : 1 mixture of the two ketone products 10 and 11 when treated with trifluoroacetic anhydride under the typical reaction conditions.5
 | ||
| Scheme 2 | ||
Because of these many problems of the synthesis especially of bridge-substituted α-[3]ferrocenophanones we considered it useful to develop a novel, conceptually unrelated synthetic route to such compounds. We have achieved this for the example of the 3-methyl substituted α-[3]ferrocenophanone system 8. The development of synthetic pathways to racemic-8 and a related route to an enantiomerically pure example of the ketone 8 will be described in this article, together with the results of the pertinent electrochemical investigation.
Results and discussion
Synthetic developments
We had recently shown that substituted [3]ferrocenophane systems can very easily be prepared by an intramolecular Mannich-type coupling reaction.10,11 Treatment of 1,1′-diacetylferrocene (12) with e.g. dimethylamine with catalytic amounts of TiCl4 rapidly results in the formation of the unsaturated [3]ferrocenophane derivative 13 (Scheme 3). Subsequent catalytic hydrogenation then gives a ca. 6 : 1 mixture of the corresponding saturated trans- and cis-[3]ferrocenophane amines from which the trans-14 isomer is obtained by recrystallization.12 Both the pure enantiomers of 14 can readily be obtained by a straightforward optical resolution procedure that gives each of the enantiomers easily in ca. 25% yield.13 The pure enantiomers of 14 were employed as starting materials for the production of a variety of chiral chelate ligands that have found use in asymmetric catalysis.14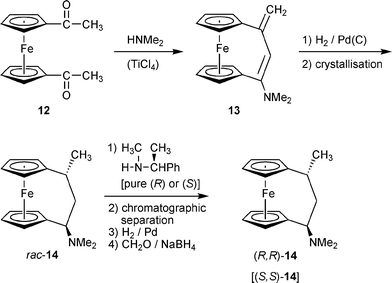 | ||
| Scheme 3 | ||
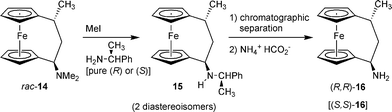
The selective transformations of the various [3]ferrocenophane derivatives were greatly facilitated by the strong neighboring group participation of the nucleophilic iron center in the SN1-type substituent exchange reactions starting from 14. Consequently, the substitution reactions at Cα of the saturated C3-bridge all proceeded strictly with overall retention,15 following a two-step reaction pathway, each of which was taking place with the usual inversion stereochemistry. In this way we have e.g. prepared the primary amino[3]ferrocenophanes (R,R)-16 and (S,S)-16 stereospecifically (Scheme 4).16
 | ||
| Scheme 4 | ||
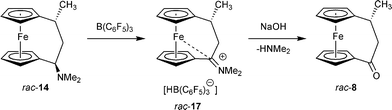
The anchimeric assistance of the Fe-atom is so pronounced that it even interacts with a strongly stabilized carbenium ion. In the [3]ferrocenophane system this was demonstrated for the iminium ion rac-17 which was formed by selective hydride abstraction from rac-14 by treatment with B(C6F5)3.17 The X-ray crystal structure analysis of the salt rac-17, supported by DFT calculations, revealed a marked Fe⋯C+ interaction even in this system (Scheme 5).
 | ||
| Scheme 5 | ||
The cation rac-17 was the first system that we have used for the synthesis of the [3]ferrocenophane ketone 8. The [3]ferrocenophane iminium salt 17 itself represents a derivative of 8, and indeed its treatment with aqueous NaOH solution led to removal of dimethylamine with formation of the ketone rac-8 (Scheme 5), that was isolated in >80% from the last hydrolytic reaction step.
Although the overall yield of rac-8 made by this procedure is acceptable and the sequence is relatively short, we thought that the use of the expensive and appearingly exotic B(C6F5)3 reagent18 might deter potential users from this principally simple synthetic pathway to the [3]ferrocenophanone 8.
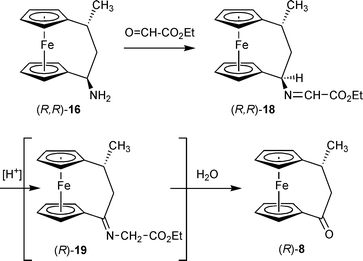
We, therefore, searched for other preparative entries. A clue was given to us by an observation made in the reaction of the primary amino[3]ferrocenophane (R,R)-16 with ethylglyoxylate. As expected, the corresponding iminoester (18) was formed. This system proved to be hydrolytically very unstable and rapidly decomposed under the applied work-up conditions to yield the [3]ferrocenophanone (R)-8 (isolated in 91% yield). Apparently, the iminoester (18) was not stable under the specific work-up conditions, but was isomerized to the functionalized imine 19. Subsequent hydrolysis of this alleged reactive intermediate probably represents a plausible mechanistic description of the observed formation of 8 (Scheme 6).
 | ||
| Scheme 6 | ||
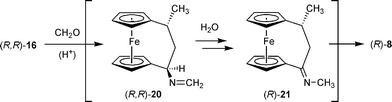
The same type of transformation can in a simpler way be carried out by treatment of the [3]ferrocenophane amine (R,R)-16 with formaldehyde. The reaction of (R,R)-16 was carried out with a 37% CH2O solution in methanol with the addition of aqueous HCl. Work-up after 30 min reflux gave the ketone (R)-8 in ca. 60% yield (see Scheme 7).
 | ||
| Scheme 7 | ||
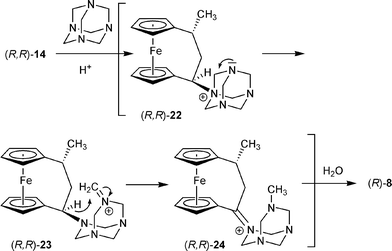
These reactions (Schemes 6 and 7) seem to represent variants of the Sommelet reaction.19 Therefore, it was tempting to try to simplify the overall reaction scheme by applying some of the reported variants of this reaction to the anticipated “benzylic” tertiary amine to ketone transformation at the [3]ferrocenophane framework. For that purpose we reacted the tertiary amine starting material (R,R)-14 with hexamethylenetetramine (urotropine) under acidic conditions. It was known from our previous work13–16 that this rapidly leads to nucleophilic exchange of the substituent at the ferrocenophane bridge α-position in a two-step process with overall retention of the configuration. We expected that the ammonium salt (R,R)-22 be formed under these conditions as a reactive intermediate that was prone to the characteristic ring opening followed by an intramolecular hydride transfer, that is characteristic of the urotropine variant of the Sommelet reaction,20 to eventually yield the corresponding ketone (R)-8 as anticipated (see Scheme 8). This was actually observed under these typical reaction conditions, and the ketone (R)-8 was isolated from the reaction mixture in 80% yield.
 | ||
| Scheme 8 | ||
It is well known that benzylic halides readily undergo this hexamethylenetetramine redox reaction to eventually yield the corresponding carbonyl products.20 Therefore, the tertiary-amine rac-14 was converted to the corresponding α-chloro[3]ferrocenophane system (rac-25) by treatment with methylchloroformate.21 As expected, the chlorination reaction proceeded with overall retention of the configuration (see Scheme 9). The subsequent hexamethylenetetramine variant of the Sommelet reaction in this case gave the [3]ferrocenophanone rac-8 in an overall yield of >90% starting from the readily available [3]ferrocenophane amine rac-14.
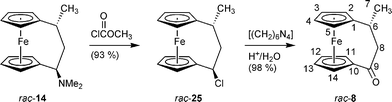 | ||
| Scheme 9 | ||
Characterization of the [3]ferrocenophanone 8
The IR spectrum of 8 shows a ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band at 1655 cm−1 (vs). In the 13C NMR spectrum the corresponding carbonyl carbon resonance was observed at δ 209.0 (in CD2Cl2). The chiral system (R)-8 features ten well separated carbon NMR resonances of the pair of η5-C5H4 ligands at iron and three resonances of the remaining carbon atoms of the methyl substituted bridge [δ 52.4 (CH2), δ 39.0 (CH), δ 22.6 (CH3)]. The framework features a total of eight methine 1H NMR signals between δ 4.89 and δ 3.98. The CH3 resonance of the substituent at the bridge occurs at δ 1.24 (d, 3J = 6.8 Hz). The strongly non-planar bridge (see below) has three hydrogens attached that give rise to a typical pattern of 1H NMR features at δ 3.21 (CH), δ 3.19 and δ 2.59 (Hax and Heq of CH2).
O) band at 1655 cm−1 (vs). In the 13C NMR spectrum the corresponding carbonyl carbon resonance was observed at δ 209.0 (in CD2Cl2). The chiral system (R)-8 features ten well separated carbon NMR resonances of the pair of η5-C5H4 ligands at iron and three resonances of the remaining carbon atoms of the methyl substituted bridge [δ 52.4 (CH2), δ 39.0 (CH), δ 22.6 (CH3)]. The framework features a total of eight methine 1H NMR signals between δ 4.89 and δ 3.98. The CH3 resonance of the substituent at the bridge occurs at δ 1.24 (d, 3J = 6.8 Hz). The strongly non-planar bridge (see below) has three hydrogens attached that give rise to a typical pattern of 1H NMR features at δ 3.21 (CH), δ 3.19 and δ 2.59 (Hax and Heq of CH2).
Complex (R)-8 was characterized by X-ray diffraction (single crystals from a cyclohexane–chloroform solution (8 : 1)). Slow crystallization of rac-8 (see above) also gave single crystals suited for the X-ray crystal structure determination. It was found that spontaneous resolution had occurred during the crystallization of rac-8 and the individual crystal analyzed was of the enantiomer (S)-8. Since the results of both X-ray crystal structure analyses were practically the same for those two structure determinations, only the data obtained from (R)-8 will be presented below.
The X-ray crystal structure analysis of (R)-8 (Fig. 1) shows an only slightly distorted ferrocene framework (Cpcentr–Fe–Cpcentr 158.1(9)°). Both the mono-substituted C5H4 ring systems are η5-coordinated to the central iron atom with the C(Cp)–Fe bond lengths being in a narrow range between 2.004(2) and 2.078(2) Å. The metallocene conformation is eclipsed. The framework of the C3-bridge is strongly folded, almost as strongly as it was previously observed for the saturated [3]ferrocenophane analogues of this series.12–14,16 The carbonyl CO vector (C9–O 1.224(2) Å) is oriented slightly out of the adjacent Cp-plane (dihedral angles: C14–C10–C9–O 41.9(3)°, C11–C10–C9–O −155.0(2)°). The carbonyl group itself is trigonally planar (sum of bond angles at C9: 359.7°) with the C10–C9–C8 angle (117.9(2)°) being slightly smaller than C10–C9–O (121.2(2)°) and C8–C9–O (120.6(2)°). The marked folding of the bridge (see Fig. 1) (C10–C9–C8–C6 82.9(2)°, O–C9–C8–C6 −90.3(2)°, C9–C8–C6–C1 −61.9(2)°) results in a strong differentiation of axial and equatorial positions at the framework. At C6 the CH3 group consequently favours an equatorial orientation (C9–C8–C6–C7 174.7(2)°).
![Two projections of the chiral framework of the [3]ferrocenophanone (R)-8.](/image/article/2006/DT/b515043d/b515043d-f1.gif) | ||
| Fig. 1 Two projections of the chiral framework of the [3]ferrocenophanone (R)-8. | ||
The electrochemical features of complex rac-8 were investigated. Its yellow CH2Cl2 solution (λmax = 436 nm) exhibits the expected one-electron oxidation to the corresponding ferrocenium monocation [8]+ (E°′ = +0.63 V, vs. SCE). Analysis of the pertinent cyclic voltammetric responses with scan rates varying from 0.02 to 2.00 V s−1 shows that: (i) the current function ipav−1/2 maintains substantially constant (a decrease by about 10% occurs upon each ten time increase of the scan rate); (ii) the current ratio ipc/ipa is constantly equal to 1; (iii) the peak-to-peak separation progressively increases from 77 to 169 mV. Such parameters feature a chemically reversible (in the short time scale of cyclic voltammetry), but electrochemically quasireversible one-electron process, which foreshadows that some structural constrain must be overcame to trigger the electron removal (or, minor structural changes accompany the oxidation).22 In confirmation of the complete chemical stability of [8]+, the green solution (λmax = 616 nm) resulting from controlled potential exhaustive one-electron oxidation (Ew = +0.8 V) displays a cyclic voltammetric profile quite complementary to the original one.
Under the same experimental conditions, monoacetylferrocene undergoes oxidation at E°′ = +0.66 V, whereas diacetylferrocene (12) oxidizes at E°′ = +0.88 V. Both the monocations are not completely stable, in that the original voltammetric signals can be only in part recovered from their exhaustively oxidized solutions. This not only means that the three-carbon bridge attenuates the electron-donating effect of the outer methyl group(s), but also tends to stabilize the ferrocenium form with respect to the vaguely reminiscent open chain analogs.
Finally, it is useful to compare the redox activity of the keto derivative 8 with that of the corresponding dimethylamino complex 14. In agreement with the basic character of the NMe2 function (vs. the acidic character of the carbon atom of the carbonyl group), complex 14 oxidizes notably easier (E°′ = +0.37 V). The corresponding monocation is not however completely stable and converts to a presently unknown new species which oxidizes reversibly at E°′ = +0.60 V.
Conclusion
We have shown that the [3]ferrocenophanone system 8 can readily be obtained on a preparative scale by non-Friedel–Crafts pathways, making use of the very convenient Mannich-type coupling reaction, starting from 1,1′-diacetylferrocene, to close the C3-bridge between the pair of Cp-rings at iron. Several pathways have been investigated and eventually variants of the Sommelet reaction were found to provide useful high yield entries to the racemic ketone rac-8 as well as the pure enantiomer (R)-8. It appears that further developments of such reaction pathways may increasingly open very attractive synthetic alternatives to the established Friedel–Crafts-type pathways for the preparation of important key compounds in ferrocene chemistry and related areas of organometallic chemistry. Complex 8 undergoes reversibly oxidation to its monocation, which, at variance with the monocations of the open-chain analog monoacetylferrocene or of the dimethylamino analog 14, which are stable only in the short times of cyclic voltammetry, is indefinitely stable.Experimental
General
All reactions were carried out in ambient atmosphere or, when mentioned, under dry argon in Schlenk-type glassware or in a glove-box. Solvents were dried and distilled prior to use. NMR spectra were recorded with a Varian UNITYplus 600 (1H: 599.9 MHz; 13C: 150.8 MHz) spectrometer. Carbon NMR spectra were recorded proton decoupled (CPD). J values are given in Hz. Atom numbering scheme used for the description of NMR spectra as in Scheme 9. Assignments were confirmed by 2D NMR spectra. Optical rotation values [α]D20 are given in 10−1 deg cm2 g−1 (c in g/100 cm3). The following instruments were used for additional physical characterization: IR spectroscopy: Nicolet 5DXC FT-IR spectrometer; melting points: DSC 2010 (TA instruments); elemental analyses: Foss Heraeus CHN-O-Rapid; optical rotation: Perkin-Elmer polarimeter 351; circular dichroism: Jobin Yvon-Spex CD-6 dichrograph. Materials and apparatus for electrochemistry have been described elsewhere.23 Potential values are referred to the Saturated Calomel Electrode (SCE). Under the actual experimental conditions (CH2Cl2 solution; [NBu4][PF6] 0.2 mol dm−3 as supporting electrolyte; platinum working electrode), the one-electron oxidation of ferrocene occurs at +0.39 V. 1,1′-Diacetylferrocene 12, optically active ferrocenophane amines 14, 16 and iminium compound 17 were prepared as previously reported.Oval), 1448s, 1099s, 1038m, 806s, 526s, 448m. [α]D20 +763 (c = 0.1 in acetonitrile). CD (c = 0.05 in acetonitrile) λmax/nm (Δε/dm3 cm−1 mol−1) = 460 (+1.47), 360 (−0.67), 331 (+3.29), 285 (+1.03), 231 (+4.15) nm. δH (599.9 MHz, CD2Cl2, 298 K) 4.89 (m, 1 H, 14-H), 4.69 (m, 1 H, 2-H), 4.67 (m, 1 H, 11-H), 4.49 (m, 1 H, 5-H), 4.37 (m, 1 H, 13-H), 4.33 (m, 1 H, 12-H), 4.09 (m, 1 H, 3-H), 3.98 (m, 1 H, 4-H), 3.21 (dqd, 3J 10.4, 6.8, 3.3, 1 H, 6-H) 3.19 (dd, 2J 9.8, 3J 10.4, 1 H, 8ax-H), 2.59 (dd, 2J 9.8, 3J 3.3, 1 H, 8eq-H), 1.24 (d, 3J 6.8, 3 H, 7-H). δC (150.8 MHz, CD2Cl2, 298 K) 209.0 (C9), 93.6 (C1), 76.7 (C10), 72.7 (C13), 72.6 (C14), 72.4 (C12), 71.6 (C5), 70.1 (C3), 68.9 (C11), 68.6 (C4), 67.8 (C2), 52.4 (C8), 39.0 (C6), 22.6 (C7). m/z (ESI+) 287.1 (23%, [M + MeOH + H]+), 277.0 (11, [M + Na]+), 255.0 (100, [M + H]+).
CH–CO2Et), 4.31–4.00 (m, 8 H, 2-H, 3-H, 4-H, 5-H, 11-H, 12-H, 13-H, 14-H), 4.28 (q, 3J 7.1, 2 H, –CH2CH3), 4.13 (dd, 3J 9.4, 2.9, 1 H, 9-H), 2.71 (qdd, 3J 7.1, 6.5, 3.6, 1 H, 6-H), 2.43 (ddd, 2J 13.3, 3J 9.4, 3.6, 1 H, 8-Hexo), 2.15 (ddd, 2J 13.3, 3J 6.5, 2.9, 1 H, 8-Hendo), 1.33 (t, 3J 7.1, 3 H, –CH2CH3), 1.24 (d, 3J 7.1, 3H, 7-H). δC (150.8 MHz, CD2Cl2, 298 K) 163.4 (CO2CH2CH3), 153.7 (–NCH–CO2Et), 92.0 (C10), 86.7 (C1), 69.5, 69.3, 69.1, 68.9, 68.5, 68.3, 67.9, 67.7 (C2, C3, C4, C5 and C11, C12, C13, C14), 63.6 (C9), 61.9 (–CO2CH2CH3), 51.5 (C6), 26.3 (C8), 18.8 (C7), 14.2 (–CO2CH2CH3). An aliquot of (R,R)-18 was dissolved in pentane and dry hydrogen chloride was bubbled through the solution. The resulting deep red precipitate was separated via filtration and analyzed by mass spectrometry. m/z (ESI+) 239.1 (20%, [M − NCH–CO2Et]+), 340.3 (100, [M + H]+). Addition of a dichloromethane solution (20 mL) containing the iminium chloride [(R,R)-18-H]+ Cl− (420 mg, 1.12 mmol) to a solution of 181 mg (1.36 mmol, 1.2 eq.) aluminium chloride in dichloromethane at −78 °C followed by quenching with ice yielded in the formation of (R)-8 (259 mg, 1.02 mmol, 91%) after work-up and purification as described above. Single crystals suitable for X-ray diffraction were grown from a saturated solution of cyclohexane–chloroform (8 : 1).
Crystallographic data for complex (R)-8
Crystal data for C14H14FeO: M = 254.10, orthorhombic, space group P212121 (no. 19), a = 5.740(1), b = 13.703(1), c = 13.774(1) Å, V = 1083.4(2) Å3, Dc = 1.558 g cm−3, µ = 1.363 mm−1, Z = 4, λ = 0.71073 Å, T = 198 K, 8802 reflections collected (±h, ±k, ±l), [(sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ)/λ] = 0.66 Å−1, 2581 independent (Rint = 0.034) and 2400 observed reflections [I
≥ 2 σ(I)], 146 refined parameters, R = 0.025, wR2 = 0.054.
θ)/λ] = 0.66 Å−1, 2581 independent (Rint = 0.034) and 2400 observed reflections [I
≥ 2 σ(I)], 146 refined parameters, R = 0.025, wR2 = 0.054.
CCDC reference number 287243.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b515043d
Acknowledgements
Financial support from the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft is gratefully acknowledged. P. Z. gratefully acknowledges the financial support from the University of Siena (PAR 2003).References
- A. Mernyi, C. Kratky, W. Weissensteiner and M. Widhalm, J. Organomet. Chem., 1996, 508, 209–218 CrossRef CAS; F. Gómez-de la Torre, F. A. Jalón, A. López-Agenjo, B. R. Manzano, A. Rodríguez, T. Sturm, W. Weissensteiner and M. Martínez-Ripoll, Organometallics, 1998, 17, 4634–4644 CrossRef CAS; F. A. Jalón, A. López-Aganjo, B. R. Manzano, M. Moreno-Lara, A. Rodríguez, T. Sturm and W. Weissensteiner, J. Chem. Soc., Dalton Trans., 1999, 4031–4039 RSC; T. Sturm, W. Weissensteiner, K. Mereiter, T. Kégl, G. Jeges, G. Petölz and L. Kollár, J. Organomet. Chem., 2000, 595, 93–101 CrossRef CAS; E. M. Caynela, L. Xiao, T. Sturm, B. R. Manzano, F. A. Jalon and W. Weissensteiner, Tetrahedron: Asymmetry, 2000, 11, 861–869 CrossRef CAS; S. Allenmark and A. Grundström, Chem. Scr., 1973, 4, 69–75 CAS; G. Tainturier, K. Chhory Sok and B. Gautheron, C. R. Acad. Sci. Paris, Ser. C, 1973, 277, 1269–1270 Search PubMed; P. Dixneuf, Tetrahedron Lett., 1971, 12, 1561–1563 CrossRef; P. Dixneuf and R. Dabard, Bull. Soc. Chim. Fr., 1972, 7, 2847–2854.
- Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, VCH, Weinheim, 1995 Search PubMed.
- W. Mock and J. R. Richards, J. Org. Chem., 1962, 27, 4050–4051 CrossRef CAS.
- K. L. Rinehart and R. J. Curby, J. Am. Chem. Soc., 1957, 79, 3290–3291 CrossRef CAS; K. L. Rinehart, R. J. Curby and P. E. Sokol, J. Am. Chem. Soc., 1957, 79, 3420–3424 CrossRef; K. L. Rinehart, R. L. Curby, D. H. Gustafson, K. G. Harrison, R. E. Bozak and D. E. Bublitz, J. Am. Chem. Soc., 1962, 84, 3263–3269 CrossRef CAS.
- M. Rosenblum, A. K. Banerjee, N. Danieli, R. W. Fish and V. Schlatter, J. Am. Chem. Soc., 1963, 85, 316–324 CrossRef CAS.
- K. L. Rinehart, B. E. Bublitz and D. H. Gustafson, J. Am. Chem. Soc., 1963, 85, 970–982 CrossRef CAS.
- Reviews: W. E. Watts, Organomet. Chem. Rev., 1967, 2, 231–254 Search PubMed; W. E. Watts, Fe organic compounds, Gmelin Handbook of Inorganic Chemistry, Springer-Verlag, Berlin, 8th edn, 1986, vol. A8 CAS; B. W. Rockett and G. Marr, J. Organomet. Chem., 1991, 416, 327–398 CAS , and preceding annual reviews by these authors.
- T. E. Turbitt and W. E. Watts, J. Organomet. Chem., 1972, 46, 109–117 CrossRef CAS.
- T. Bitterwolf, Inorg. Chim. Acta, 1986, 117, 55–64 CrossRef CAS.
- S. Knüppel, R. Fröhlich and G. Erker, J. Organomet. Chem., 1999, 586, 218–222 CrossRef CAS; S. Knüppel, R. Fröhlich and G. Erker, J. Organomet. Chem., 2000, 595, 307–312 CAS.
- S. Knüppel, G. Erker and R. Fröhlich, Angew. Chem., 1999, 111, 2048–2051 CrossRef; S. Knüppel, G. Erker and R. Fröhlich, Angew. Chem., Int. Ed., 1999, 38, 1923–1926 CrossRef CAS; S.-D. Bai, X.-H. Wei, J.-P. Guo, D.-S. Liu and Z.-Y. Zhou, Angew. Chem., 1999, 111, 2051–2054 CrossRef; S.-D. Bai, X.-H. Wei, J.-P. Guo, D.-S. Liu and Z.-Y. Zhou, Angew. Chem., Int. Ed., 1999, 38, 1926–1928 CrossRef CAS.
- P. Liptau, S. Knüppel, G. Kehr, O. Kataeva, R. Fröhlich and G. Erker, J. Organomet. Chem., 2001, 637–639, 621–630 CrossRef CAS.
- P. Liptau, L. Tebben, G. Kehr, B. Wibbeling, R. Fröhlich and G. Erker, Eur. J. Inorg. Chem., 2003, 19, 3590–3600 CrossRef; P. Liptau, L. Tebben, G. Kehr, B. Wibbeling, R. Fröhlich and G. Erker, Eur. J. Inorg. Chem., 2003, 19, 4261 CrossRef.
- P. Liptau, T. Seki, G. Kehr, A. Abele, R. Fröhlich, G. Erker and S. Grimme, Organometallics, 2003, 22, 2226–2232 CrossRef CAS; P. Liptau, D. Carmona, L. A. Oro, F. J. Lahoz, G. Kehr and G. Erker, Eur. J. Inorg. Chem., 2004, 23, 4586–4590 CrossRef; P. Liptau, L. Tebben, G. Kehr, R. Fröhlich, G. Erker, F. Hollmann and B. Rieger, Eur. J. Org. Chem., 2005, 9, 1909–1918 CrossRef.
- D. Marquarding, H. Klusacek, G. Gokel, P. Hoffmann and I. Ugi, J. Am. Chem. Soc., 1970, 92, 5389–5393 CrossRef CAS; G. Gokel, D. Marquarding and I. Ugi, J. Org. Chem., 1972, 37, 3052–3058 CrossRef CAS; T. Hayashi, T. Mise, M. Fukushima, M. Kagotani, N. Nagashima, Y. Hamada, A. Matsumoto, S. Kawakami, M. Konishi, K. Yamamoto and M. Kumada, Bull. Chem. Soc. Jpn., 1980, 53, 1138–1151 CAS; C. Ganter and T. Wagner, Chem. Ber., 1995, 128, 1157–1161 CrossRef CAS.
- L. Tebben, G. Kehr, R. Fröhlich and G. Erker, Synthesis, 2004, 12, 1971–1976.
- P. Liptau, M. Neumann, G. Erker, G. Kehr, R. Fröhlich and S. Grimme, Organometallics, 2004, 23, 21–25 CrossRef CAS.
- G. Erker, Dalton Trans., 2005, 1883–1890 RSC.
- M. Sommelet, Bull. Soc. Chim. Fr., 1918, 23, 95–97; O. Bayer, in, Houben–Weyl: Methoden der Organischen Chemie, Thieme, Stuttgart, 1954, vol. VII/I, p. 194 Search PubMed; S. H. Dandegaonker, Ind. J. Appl. Chem., 1966, 29, 91–94 Search PubMed; Y. Tang, B. Cheng and T. Hu, Zhongman Gongye Daxue Xuebao, 1995, 26, 527–531 Search PubMed.
- S. N. Kilenyi, in Encyclopedia of Reagents for Organic Synthesis, ed. L. A. Paquette, Wiley, New York, 1995, vol. 3, p. 2666 Search PubMed; N. Blazevic, D. Kolbah, B. Belin, V. Sunjic and F. Kajfez, Synthesis, 1979, 161–179 Search PubMed.
- I. Weber and G. B. Jones, Tetrahedron Lett., 2001, 42, 6983–6986 CrossRef CAS; U. Englert, A. Salzer and D. Vasen, Tetrahedron: Asymmetry, 1998, 9, 1867–1870 CrossRef CAS; U. Englert, R. Harter, D. Vasen and A. Salzer, Organometallics, 1998, 18, 4390–4398 CrossRef CAS.
- P. Zanello, in Inorganic Electrochemistry. Theory, Practice and Application, RSC, Cambridge, 2003 Search PubMed.
- F. Fabrizi de Biani, M. Corsini, P. Zanello, H. Yao, M. E. Bluhm and R. N. Grimes, J. Am. Chem. Soc., 2004, 126, 11360–11369 CrossRef CAS.
Footnotes |
| † X-Ray crystal structure analysis. |
| ‡ Electrochemistry. |
| This journal is © The Royal Society of Chemistry 2006 |