Coordination chemistry of phosphanyl amino acids: solid state and solution structures of neutral and cationic rhodium complexes
Christian
Meyer
a,
Markus
Scherer
a,
Hartmut
Schönberg
a,
Heinz
Rüegger
a,
Sandra
Loss
a,
Volker
Gramlich
b and
Hansjörg
Grützmacher
*a
aLaboratory of Inorganic Chemistry, Department of Chemistry and Applied Biosciences, ETH Hönggerberg, HCI H131, CH-8093, Zürich, Switzerland. E-mail: gruetzmacher@inorg.chem.ethz.ch; Fax: int. +41 1 632 1032
bLaboratory of Crystallography, Wolfgang-Pauli-Str. 10, ETH Hönggerberg, HCI G 503, CH-8093, Zürich, Switzerland
First published on 29th November 2005
Abstract
Copper phosphide or arsenide complexes, [Cu(EPh2)(neo)] (E = P, As, neo = 2,9-dimethyl-1,10-phenanthroline; trivial name: neocuprine) react selectively with the N-protected brominated serine derivatives, 2-(S)-(alkoxycarbonylamino)-3-bromomethylpropionates 1a–c (ROCOSerBr, a: R = PhCH2, b: tBu, c: Me) to give the corresponding phosphanylated or arsanylated amino acids, ROCOSerPhos (3a–c: Phos = PPh2) and ZSerArs 7 (Ars = AsPh2, Z = PhCH2OCO). The dipeptide ZAlaSerPhos 3d was likewise prepared. The phosphanes 3a–d, and the arsane 7 reacted cleanly with [Rh2(µ-Cl)2(cod)2] to give the rhodium(I) complexes [RhCl(cod)(ZSerPhos)] 8, [RhCl(cod)(BocSerPhos)] 9 (Boc = tBuOCO), [RhCl(cod)(ZAlaSerPhos)] 10, and [RhCl(cod)(ZSerArs)] 11 which were characterized by X-ray diffraction studies. A common structural feature is an intramolecular (N)H⋯Cl(Rh)-hydrogen bridge which according to NMR investigations remains intact in solution. The abstraction of chloride from the coordination sphere of Rh(I) in 8 or 10 has a profound structural impact. While in 8 and 10, the ligands bind in a monodentate fashion, via the phosphorus atom only, they serve as bidentate ligands via the phosphorus centre and the peptidic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in [Rh(cod)(κ2-ZSerPhos)]PF612 and [Rh(cod)(κ2-ZAlaSerPhos)]PF613. This causes also the amino acid residue structures to change from α-helix type in 8 and 10 to a β-sheet type in 12 and 13. Addition of chloride to 12 and 13 fully re-establishes the structures of 8 and 10. The complexes [RhCl(cod)(ZSerPhos)] 8 and [RhCl(cod)(BocSerPhos)] 9 show good activities in homogeneously catalyzed hydrogenations of olefins while the dipeptide complex 10 is less active. Phosphane addition to 8 greatly diminishes the catalytic activity. The cationic complex [Rh(cod)(κ2-ZAlaSerPhos)]PF6 shows low activity which, however, is greatly increased by addition of one equivalent of phosphane.
O group in [Rh(cod)(κ2-ZSerPhos)]PF612 and [Rh(cod)(κ2-ZAlaSerPhos)]PF613. This causes also the amino acid residue structures to change from α-helix type in 8 and 10 to a β-sheet type in 12 and 13. Addition of chloride to 12 and 13 fully re-establishes the structures of 8 and 10. The complexes [RhCl(cod)(ZSerPhos)] 8 and [RhCl(cod)(BocSerPhos)] 9 show good activities in homogeneously catalyzed hydrogenations of olefins while the dipeptide complex 10 is less active. Phosphane addition to 8 greatly diminishes the catalytic activity. The cationic complex [Rh(cod)(κ2-ZAlaSerPhos)]PF6 shows low activity which, however, is greatly increased by addition of one equivalent of phosphane.
1 Introduction
It is an attractive goal to incorporate the complexity of natural structures into ligands for transition metal complexes used in homogeneous catalysis.1 Carbohydrates, peptides and proteins offer such structural diversity and hence are potential targets for further functionalization. In order to enhance the stability of the catalytic entities one needs to bind tightly the catalytically active late transition metal centres (i.e. Rh, Ir, Pd, Pt, etc.) and for that purpose non-natural donor centres may be introduced into the natural ligand framework. Among these, phosphanyl groups, R2P–, are immediately evident because many phosphane complexes proved to be active catalysts (or precursors to such),2 phosphanes show sufficiently high binding constants to metals,3 and phosphanyl groups itself can be sterically and electronically tuned via different substituents R. Phosphanes with carbohydrate backbones are meanwhile firmly established as rather easily accessible ligands in enantioselective catalysis.4 Phosphanyl-substituted peptides have been investigated in the last years mainly by Gilbertson and co-workers as ligands in homogeneously transition metal catalyzed reactions.1 Especially, serine5 and proline6 based phosphanes proved to be suitable for this purpose and for the parallel syntheses of large ligand libraries.1,7 The (phosphanyl)polypeptides were transformed into rhodium complexes and tested in catalytic hydrogenations. In some cases these experiments were performed with the catalyst precursors still attached to the support used for polypeptide synthesis. A further interesting development in this area is the possibility to incorporate the (phosphanyl)peptides in larger aggregates with some sort of tertiary structures.7a This idea has been tested with some success with (phosphanyl)carbohydrates organized on the surface of micelles which led to an increase of the enantioselectivity in catalytic hydrogenations.4c The attachment of achiral rhodium complexes to chiral carbohydrate or peptide amphiphiles is particularly interesting, but had little success so far.8Some structural data for bis(phosphanyl)amino peptides and their complexes were obtained from NMR spectroscopic studies in solution and the structure of a dodecamer containing two (diphenylphosphano)serine (SerPhos) units in internal positions i and i + 4 could be determined by X-ray analyses.9 However, many fundamental aspects of the coordination chemistry of phosphanyl peptides remain to be investigated. In this paper we report: (i) the details of the synthesis of phosphanyl- and arsanyl-substituted amino acids using a method developed in our laboratory;10 (ii) the X-ray structure analyses of (phosphanyl)- and (arsanyl)-amino acid metal complexes, i.e. [RhCl(RSerPhos) and [RhCl(RSerArs)] with R = substituent at the N-terminus of the coordinated amino acid; (iii) a comparison of the solid state structures with the corresponding structures in solution (determined by NMR) and, specifically, how changes in the coordination sphere of the transition metal centre cause significant structural changes of the peptide residues. (vi) Finally, we report results concerning the performance of these complexes as precursors in catalytic hydrogenations.
2 Results and discussion
Syntheses
In contrast to Gilbertson's approach consisting in synthesizing a small phosphanyl-substituted amino acid building block which is used in subsequent coupling reactions in order to obtain larger peptides, we thought it might be an equally valuable alternative to find a selective reagent which allows the introduction of the phosphanyl group into the peptide as disclosing step of ligand synthesis. Indeed, copper phosphide complexes with heterocyclic nitrogen ligands like 2 are such reagents and allow the selective replacement of halogenide functionalities for Ph2P groups.10 Other functional groups as carbonyl groups do not interfere (Scheme 1). Thus, the 3-bromo-2-N-carboxymethylpropionate derivatives 1a–c derived from serine react smoothly at room temperature with 2 under Br/Ph2P exchange to give the phosphanyl amino acid copper complexes [CuBr(neo)(3a–c)] as orange–yellow substances (neo = 2,9′-dimethylphenanthroline).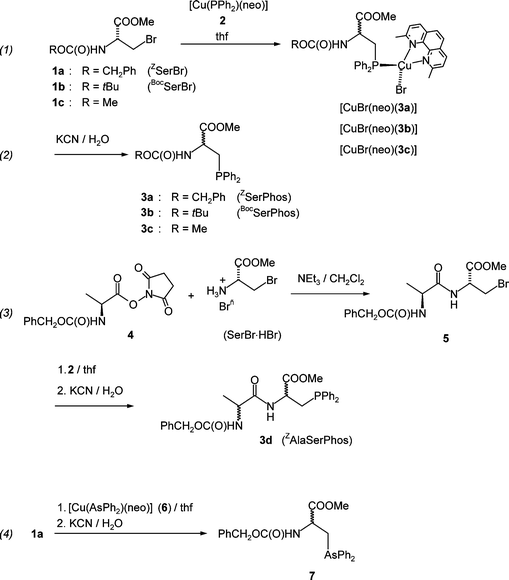 | ||
| Scheme 1 Syntheses of ROCOSerPhos derivatives 3 [a: R = CH2Ph (ZSerPhos), b: R = tBu (BocSerPhos), c: R = Me), ZAlaSerPhos 3d and ZSerArs 7. | ||
As N-protecting groups, we used N-benzyloxy carbonyl, PhCH2OCO = Z, N-tert-butoxycarbonyl, tBuOCO = Boc and N-methoxycarbonyl, MeOCO. The copper complexes can be isolated and [CuBr(neo)(3c)] was obtained in high yield (87%). Usually, however, the reaction mixtures were treated with KCN/H2O in order to deliberate the free phosphanyl serine derivatives 3a–c in about 70% yields.
Starting from the Z-L-alanine hydroxysuccinimide ester 4 and (R)-2-amino-3-bromomethylpropionate hydrobromide (SerBr HBr), the dipeptide 5 was obtained in about 50% yield. Reaction with 2 gave, after work-up with aqueous KCN the desired ligand ZAlaSerphos 3d in good isolated yield (∼70%). In an analogous way, the arsanyl substituted serine derivative 7 (SerArs) is obtained when the copper arsenide complex [CuAsPh2(neo)] 6 is used in the reaction with 1a. Unfortunately, the reactions proceed under racemization of the stereogenic centres which is a serious drawback of our method. While our work was in progress, Stelzer and co-workers reported a facile synthesis of SerPhos from N-Boc-3-iodo-L-alanine and Ph2PH in DMF using K2CO3 as base.11 However, this reaction proceeds also under racemization. Only Gilbertson and co-workers reported a short and elegant stereospecific synthesis of the P-sulfide of SerPhos using a copper/zinc reagent made from a commercially available iodo amino acid.12
Reaction of the phosphanes 3a,b,d or arsane 7 with [Rh2(µ-Cl)2(cod)2] in ethanol gave the complexes 8, 9, 10 and 11, respectively, in good yields (Scheme 2). All complexes were obtained in high yield as yellow to orange crystals after re-crystallization from saturated acetonitrile solutions. In the solid state, 8–11 are quite stable and can be handled on air.
 | ||
| Scheme 2 Syntheses of rhodium(I) complexes 8–11 with ZSerPhos 3a, BocSerPhos 3b, ZAlaSerPhos 3d and ZSerArs 7 as ligands. | ||
Structures in the solid state
The single crystals contain the racemates of 8, 9 and 11 in each case and diastereomers of 10. One of the stereoisomers of 9, 10 and 11 is depicted in Fig. 1(A), (B) and (C). The ZSerphos complex 8 has a structure very similar to the one of the arsenic analogue 11 and is therefore not especially shown in Fig. 1. Selected bond angles and distances of all complexes are compiled in Table 1 and details on the data collection and refinement are given in Table 6 in the Experimental section.| Cβ–Cα–N | P–Cβ–Cα | Rh–P(As)–Cβ | Cl–Rh–P(As) | |||
|---|---|---|---|---|---|---|
| a The average of the two independent molecules is given which have very similar structures. b The bridging hydrogen bonds c (NH⋯Cl) were calculated from the experimentally determined N⋯Cl distances assuming a NH distance of 0.9 Å. | ||||||
| 8 a | 112.0(7) | 117.4(6) | 118.1(3) | 88.6(8) | ||
| 9 | 110.5(4) | 115.7(3) | 110.2 (1) | 86.70(5) | ||
| 10 | 111.7(3) | 117.7(3) | 118.7(1) | 90.13(4) | ||
| 11 | 106.8(5) | 116.5(4) | 120.3(2) | 88.60(4) | ||
![Structure plots of R-[RhCl(cod)(BocSerPhos)] R-9 (A), R,S-[RhCl(cod)(ZAlaSerPhos)] R,S-10 (B) and R-[RhCl(cod)(ZSerArs)] R-11 (C). Thermal ellipsoids are shown at 30% probability. Selected bond lengths (Å) and angles (°) are listed in Table 1.](/image/article/2006/DT/b512653c/b512653c-f1.gif) | ||
| Fig. 1 Structure plots of R-[RhCl(cod)(BocSerPhos)] R-9 (A), R,S-[RhCl(cod)(ZAlaSerPhos)] R,S-10 (B) and R-[RhCl(cod)(ZSerArs)] R-11 (C). Thermal ellipsoids are shown at 30% probability. Selected bond lengths (Å) and angles (°) are listed in Table 1. | ||
For the BocSerphos complex 9 (Fig. 1(A)) and the ZSerArs complex 11 (Fig. 1(C)), respectively, the enantiomer with the natural R-configuration at the α-serine carbon atom is shown. For the dipeptide complex 10 (Fig. 1(B)), the stereoisomer with the non-natural S-configuration at the α-carbon of the serine residue is presented. In this compound, the S-stereogenic centre in the alanine residue, Ala, appears to be disordered as a consequence of crystal packing.
The most remarkable and common feature in the structures of 8–11 is the N–H⋯Cl bridge which leads to the formation of a distorted seven-membered ring including the Rh, P, Cβ, Cα, N, H and Cl centres. The distance c between the N-hydrogen atom and the rhodium-coordinated chlorine atom varies between 2.18 Å (9) and 2.62 Å (8) and the N–H⋯Cl angle varies by about 7° from 153° (8) to 160° (9). The N–H⋯Cl bridges lead to an arrangement of the N-terminal protecting groups, Z or Boc, above one side of the square planar coordination sphere of the rhodium centres. The deviation from planarity given by the intersection φ of the plane running through Rh and the midpoints of the coordinated CCcod bonds with the plane running through Rh, P and Cl is small for all complexes: 8: φ = 13°, 9, 11 = 6°. All other structural data show values in the expected ranges and do not differ much within series 8–11. The phenyl groups bonded to phosphorus show the typical edge-to-face arrangements with interplane angles of 50–70°.4g
In Table 2, the torsion angles Θa1, Θa2 and Θb (and Θh, Θj for 10) are listed which we will use in the following to discuss the coordination spheres created by the phosphanyl substituted amino acids. The corresponding values of the antipodes are simply obtained by multiplying the given data by minus one. As examples, Newman projections along the bonds CβCα = a and CαN = b, are presented for the R-configured SerPhos unit in 9 in Fig. 2(A). For the dipeptide complex 10, projections for the R,S- and S,S-configured ZAlaSerPhos moieties are presented and the torsion angles along a and b and h and j are given. The conformations for the ZSerPhos and ZSerArs complexes 8 and 11, respectively, resemble closely the one presented for R,S-10.
| Θ a 1 | Θ a 2 | Θ b | ϕ Serphos | Θ h | ψ Ala | Θ j | ϕ Ala | |
|---|---|---|---|---|---|---|---|---|
| a Average of two independent molecules per unit cell. | ||||||||
| R-8a | −52.2 | −168.0 | −139.3 | −79 | — | — | — | |
| R-9 | −81.3 | 163.0 | −120 | −60 | — | — | — | |
| R,S-10 | −55.7 | −171.2 | −130.5 | −70 | 31.1 | −28.9 | −127.7 | −67.7 |
| R-11 | −50.6 | −166.6 | −159.9 | −100 | — | — | — | |
| S,S-10 | 55.7 | 171.2 | 130.5 | 70 | 110.3 | 50.3 | −13.7 | 46.3 |
![(A) Newman projections along bonds a and b in the Serphos unit in R-[RhCl(cod)(BocSerPhos)] (R-9). (B) and (C): Newman projections along bonds a, b, c and d in the AlaSerphos unit in the diastereomers R,S-[RhCl(cod)(ZAlaSerPhos)] (R,S-10) and S,S-[RhCl(cod)(ZAlaSerPhos)] (S,S-10). The torsion angles Θa1, Θa2, Θb, Θh and Θj. are indicated and the corresponding data are listed in Table 2. (D) Schematic presentation of the seven-membered chelate ring in 8–11.](/image/article/2006/DT/b512653c/b512653c-f2.gif) | ||
| Fig. 2 (A) Newman projections along bonds a and b in the Serphos unit in R-[RhCl(cod)(BocSerPhos)] (R-9). (B) and (C): Newman projections along bonds a, b, c and d in the AlaSerphos unit in the diastereomers R,S-[RhCl(cod)(ZAlaSerPhos)] (R,S-10) and S,S-[RhCl(cod)(ZAlaSerPhos)] (S,S-10). The torsion angles Θa1, Θa2, Θb, Θh and Θj. are indicated and the corresponding data are listed in Table 2. (D) Schematic presentation of the seven-membered chelate ring in 8–11. | ||
With the exception of complex 9, similar torsion angles Θa1 and Θa2 are observed which define the position of the Ph2PCH2-side chain vs. the stereogenic centre Cα of the phosphanyl substituted serine residue. The angle Θb defining the orientation of the planar amide NHCO unit vs. Cα varies over a broader range from Θb(min.) −120.0 in 9 to Θb(max) −159.9 in 11 and indicates conformational flexibility. Commonly, the mutual orientation of the peptide planes is defined by the dihedral angles ϕ along the N–Cα bond b and ψ along the Cα–CO bond. With the relations for α-helical conformations, ϕSerphos = Θb + 60 and ϕAla = Θj + 60, the angles ϕ were calculated and listed in Table 2 for the R-configured isomers. For compound 10, the angle ψAla = Θh
− 60 is given for the R,S-diastereomer (entry 3) and S,S-isomer (entry 5).
In the IR spectra of 8–11, a shift of the N–H stretching vibration to smaller wavenumbers by more than 100 cm−1 indicates the presence of the N–H⋯Cl bridge. Two characteristic amide modes I, II are observed in 8–11 [νI(CONH) ≈ 1700 cm−1 and νII(CONH) ≈ 1500 cm−1] and these are not significantly different from the ones seen in the free ligands (see Table 4 below).
Structures in solution
Coupling constants are frequently used to determine the mutual orientation given by ϕ and ψ of the individual peptide units in polypeptides, –CO–NH–CαHα(CβH2R)–C′O–N′H–, where Hα represents the hydrogen linked to the chiral Cα carbon of the amino acid residue, HN indicates the amide proton and Cβ is the first carbon centre of the side chain (see also Fig. 1).13 Additionally, NOE experiments can be performed to obtain more structural information. We used the 3J(Hα,HN), 3J(Hα,Hβ1) and 3J(Hα,Hβ2) coupling constants in combination with the Karplus correlation in order to determine Θa1, Θa2 and Θb in solution. The resulting data for the racemates of the uncomplexed phosphanyl amino acid 3c, the dipeptide 3d and the complexes 8–10 are given in Table 3. The resonances for the R,S- and S,S-diastereomers of 3d could be partly distinguished in the NMR spectra of the complexes 10 (but not for the free ligand) and these are denominated as 10diastereomer_1 and 10diastereomer_2. A more precise assignment of the stereochemistry cannot be made.| 3 J(Hα,Hβ1)/Hz | Θ a 1/° | 3 J(Hα,Hβ2)/Hz | Θ a 2/° | 3 J(Hα,HN)/Hz | Θ b/° | ϕ Serphos/° | |
|---|---|---|---|---|---|---|---|
| 3c | 7.7 | ±150 | ±90 | ||||
| 3d | 7.3 | ±140 | ±80 | ||||
| 8 | 3.0 | ±55 | 12.5 | ±170 | 7.5 | ±155 | ±95 |
| 9 | 3.5 | ±70 | 12.5 | ±170 | 7.4 | ±145 | ±85 |
| 10 diasteromer_1 | 2.8 | ±60 | 13.0 | ±170 | 6.8 | ±140 | ±80 |
| 10 diasteromer_2 | 3.0 | ±55 | 12.0 | ±160 | 6.8 | ±140 | ±80 |
Unfortunately, all 1H NMR spectra of solutions containing the mixture of the diastereomers of compound 10 could not be sufficiently resolved and hence we were unable to determine the torsion angles for the alanine residue in 3d and 10.
A comparison of the data listed in Tables 2 and 3, respectively, shows that the structures of the central seven-membered rhodium chelates are very similar in the solid state and in solution. The values for the torsion angles ϕSerphos (|60|°–|100|° in the solid state, |80|°–|95|° in solution) compare reasonably well with the ones determined for dodecapeptides containing two SerPhos units in i and i + 4 positions (−73° in the solid, −60 to −80° in solution).9 These values fall within the range of −60 to −90° typically found in helical conformations of peptides.14 Importantly, we assume that the N–H⋯Cl bridge is conserved in solution as is clearly indicated by the significant high-frequency shifts (>1 ppm) of the HN resonances of the SerPhos units in complexes 8 [δ(HN) 7.18], 9 [δ(HN) 6.82], 10 [δ(HN) 8.46/8.16] and 11 [δ(HN) 6.98] when compared to the corresponding free ligands 3a [δ(HN) 5.48], 3b, [δ(HN) 5.77], 3d [δ(HN) 5.8] and 7 [δ(HN) 5.51]. Such large coordination shifts were not observed with the larger peptides where the (i, i + 4)-bis(serphos)peptide binds via the two phosphane residues to a cationic rhodium norbornadiene fragment.9
Chloride abstraction reactions from complexes 8 and 10
How will the cleavage of the N–H⋯Cl bridges affect the structure of the ligand? To answer that question, the metal bonded chloride was exchanged for a weakly coordinating anion and 8 and 10 were reacted with TlPF6 in toluene. The reactions are quantitative and the products 12 and 13 were obtained as yellow powders (Scheme 3). Complex 12 can also be prepared with AgPF6 as reagent but the dipeptide complex 10 decomposes.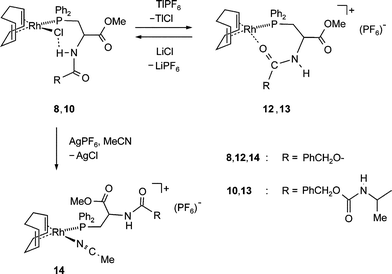 | ||
| Scheme 3 Chloride abstractions from 8 or 10 with TlPF6 to give 12 or 13, respectively. Reaction of 8 with AgPF6 in MeCN gives 14. | ||
That significant structural changes occur when the chlorine atom is removed from the coordination sphere of the rhodium atom is indicated by the change of some characteristic NMR and IR data of compounds 12–14 (see Table 4).
| δ(1H) | ||||||
|---|---|---|---|---|---|---|
| a Broadened bands in the range 3450–3380 cm−1, 1750–1710 cm−1 due to overlapping absorptions which were not assigned. b Solvents: 3a,d: C6D6, 8: CD3CN, 9: CDCl3, 10: CD2Cl2, 12: CDCl3, 13: CD2Cl2. c The resonances for the COOMe and CONH groups overlap. d The average of the signals for both diastereomers is given which are very close. | ||||||
| HN (SerPhos) | 5.48 | 5.8 | 6.98 | 8.46/8.16 | 6.26 | 8.24/8.10 |
Unfortunately, none of the products gave suitable crystals for an X-ray analysis, however, reasonable structures for the coordination sphere around the rhodium centre can be proposed. Three different coordination spheres may be assumed for 12 and 13. (a) The NH group of the amide coordinates to Rh, (b) the CO unit of the COOMe group binds to Rh, or (c) the CO group of the carbobenzoxy or alanyl unit, respectively, is bonded. The first two coordination modes give rise to six-membered the third one to a seven-membered chelate ring. Possibility (a) is rather unlikely because NH units of amides are generally weakly coordinating, no 103Rh15N coupling is observed and ν(NH) is not shifted to smaller and ν(CO) not to higher wavenumbers as would have been expected. Also possibility (b) is unlikely because all spectroscopic data [ν(CO) and δ(13C)] of the ester group remain unaffected by the chloride abstraction. We propose that 12 and 13 have the seven-membered ring structures as shown in Scheme 3 on the basis of the following observations: (i) both, ν(NH) and ν(CO) are shifted to lower wavenumbers in 12 and 13 when compared to the rhodium chloride complexes 8 and 10; (ii) no amide II absorption band is observed in 12 and 13 which is characteristic for cyclic amides, (iii) the δ(13C) resonance of the CONH group which is supposed to be involved in the coordination is slightly shifted to higher frequencies by 3–7 ppm and (iv) importantly, the 2J(103Rh13C) coupling (1.4 Hz) could be resolved for the carbon nucleus of the amide group in 12. Similar observations were made for comparable rhodium(I) complexes with seven-membered P,(CO)-chelate rings.15
Further information about the structures of 12 could be extracted from the sufficiently resolved 3J(Hα,HN) 3J(Hα,Hβ1) and 3J(Hα,Hβ2) coupling constants (8.3, 3.5 and 12.5 Hz, respectively). This results in Θb
≈
±150°, Θa1
≈
±70° and Θa2
≈
±170 which define the torsion along the CαN bond, b, and the CβCα bond, a, respectively. In contrast to the rhodium chloride complex 8, the methylene protons of the CH2PPh2 group show only one cross-peak with the phenyl protons in the NOESY spectrum. This indicates that the CH2 group has a symmetric and hence eclipsed orientation to the phenyls. With these data, a structure model for the coordination sphere of the rhodium centre in 12 can be constructed and this is shown in Fig. 3(B). Fig. 3(A) shows the NMR structure of 8 in solution for comparison. While the conformation of the SerPhos unit in 8 has a structure resembling the one of serine residues in α-helical peptides, its conformation in 12 is closer to that one in a β-sheet. As in these, an amide proton, HN, and a CO group point to the outside of the molecule. This yet unexploited feature would make intermolecular donor–acceptor interactions possible. For 13, the NMR spectra are quite complex because in addition to the doubling of all signals due to the presence of two diastereomers, the fluxional behaviour of the cyclooctadiene ligand leads to line broadening and overlap of some signals. While the 31P NMR spectra at room temperature show two doublets [1J(31P103Rh) ≈ 152 Hz], one for each diastereomer, lowering the temperature to 200 K leads to a (reversible) splitting into four doublets. Also the 1H NOESY spectra indicate that the alanine residue adopts at least two conformations. This is in contrast to the neutral rhodium chloride complex 10, where such phenomena were not observed. This finding indicates that the ZAlaSerPhos ligand 3d shows a higher flexibility in the cationic complex 13.16 Because of the almost identical characteristic bands in the IR spectra for both cationic complexes, 12 and 13, and the similar 3J(Hα,HN) coupling constants (7.1 Hz in 13), we assume rather similar conformations of the ligands within the rhodium coordination sphere as well.
![Proposed structures for the [RhCl(ZSerPhos)] fragment in 8 (A) and the [Rh(ZSerPhos)]+-fragment in 12 (B) in solution based on NMR data.](/image/article/2006/DT/b512653c/b512653c-f3.gif) | ||
| Fig. 3 Proposed structures for the [RhCl(ZSerPhos)] fragment in 8 (A) and the [Rh(ZSerPhos)]+-fragment in 12 (B) in solution based on NMR data. | ||
Importantly, when solution of 12 and 13 are treated with lithium chloride, the structural changes caused by the chloride abstraction are reversed and the spectra of the neutral chloro complexes 8 and 10 are fully recovered (Scheme 3).
When the chloro substituent in 10 is abstracted with [Ag(MeCN)2]PF6 in acetonitrile, the cationic acetonitrile complex 14 is obtained as yellow powder after evaporation of the solvent. Equally, 14 results when 12 is dissolved in MeCN. That the coordinating CO group in 12 is replaced by a MeCN in 14 is evident from the spectroscopic data: The NH stretching vibration is observed at 3400 cm−1, two amide bands for the CONH group are observed at 1708 and 1503 cm−1, and the δ(13C) resonance (156.5 ppm) equals almost the one in 3a and 8.
Catalytic hydrogenations with rhodium SerPhos complexes
The reactivity of the neutral chloro rhodium complexes 8, 9, 10 and 11 and the cationic complex 12 as catalyst precursors in homogeneously catalyzed hydrogenations was tested. For comparison, reactions with [RhCl(PPh3)3] and [RhCl(cod)(PPh3)] were included in the investigation. All reactions were performed in EtOH as solvent with 0.1 mol% catalyst at room temperature under about 4.5 bar H2 pressure. After two hours, the reaction mixtures were analyzed by gas chromatography (GC). As substrates, the olefins cyclohexene (ch), 1-hexene (1-h), acrylic acid methylester (am) and 2,3,-dimethyl-2-butene (dmb) were employed. The results are compiled in Table 5. It is generally assumed that monophosphane rhodium(I) complexes are not very reactive in catalytic hydrogenations2 and by comparison of entries 1 [catalyst source: RhCl(cod)(PPh3)] and 2 [catalyst source: [RhCl(PPh3)3] this is confirmed under our conditions. Interestingly, however, the monophosphane complex 8 (entry 3) shows a significant higher activity than the classical Wilkinson-catalyst [RhCl(PPh3)3]. As entry 4 shows, the activity is not dependent on the co-ligand in the pre-catalyst; that is the complex [RhCl(C2H4)2(ZSerPhos)] (prepared in situ) shows the same activity as 8 indicating the absence of any significant induction phase. The highest reactivity is found with complex 9 containing the Boc-protected SerPhos ligand 3b which is the only one who gives detectable amounts of 2,3-dimethylbutane as hydrogenation product of the tetrasubstituted olefin dmb. On the other hand, the dipeptide complex [RhCl(cod)(ZAlaSerPhos)] (10) (entry 6) shows the lowest activity of the neutral phosphane complexes. The arsane complex 11 is inactive (entry 7).| Conversion (%) | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst precursor | t/h | ch | 1-h | am | dmb |
| a After 18 h the following yields were obtained: [RhCl(PPh3)3]: 40 (ch), 90 (1-h), 100 (am), 0 (dmb); 8: 100 (ch), 100 (1-h), 100 (am), 0 (dmb). b Prepared in situ from equimolar amounts of ligand 3a and [Rh2(µ-Cl)2(C2H4)]. | ||||||
| 1 | [RhCl(cod)(PPh3)] | 2 | 4 | 20 | 0 | 0 |
| 2 | [RhCl(PPh3)3]a | 2 | 25 | 86 | 100 | 0 |
| 3 | [RhCl(cod)(ZSerPhos)] (8)a | 2 | 50 | 100 | 100 | 0 |
| 4 | [RhCl(C2H4)2(ZSerPhos)]b | 2 | 55 | 100 | 100 | 0 |
| 5 | [RhCl(cod)(BocSerPhos)] (9) | 2 | 92 | 100 | 100 | 5 |
| 6 | [RhCl(cod)(ZAlaSerPhos)] (10) | 2 | 1 | 10 | 12.6 | 0 |
| 7 | [RhCl(cod)(ZSerArs)] (11) | 2 | 0 | 1 | 0 | 0 |
| 8 | [Rh(cod)(ZSerPhos)](PF6) (12) | 2 | 13.8 | 4.6 | 4.3 | 0 |
| 9 | 8 + 1 eq. PPh3 | 2 | 0.6 | 5.3 | 9.1 | 0 |
| 10 | 12 + 1 eq. PPh3 | 2 | 50 | 100 | 100 | 0 |
Although most often the activity of cationic rhodium(I) complexes with weakly coordinating anions (CF3SO3, BF4, PF6, etc.) is higher than with the neutral chloro complexes, this is not true for 12 (entry 8). The catalyst derived from this complex is much less active than the one obtained with 8, especially against the alkene 1-h and ester am. Remarkably, the activity of the neutral complex 8 drops dramatically when one equivalent PPh3 is added (entry 9). On the other hand, addition of one equivalent PPh3 to the cationic complex 12 increases the activity which becomes comparable to the one of the neutral complexes 8 and [RhCl(C2H4)2(ZSerPhos)] (entry 10).
We interpret these results as follows: (1) We assume that the seven-membered Rh–P–C–C–N–H⋯Cl ring remains intact and the solvated [RhCl(RSerPhos)] fragment is the catalytically active species in reactions with 8, 9 and 10. Note, that the group R is orientated above the metal center in the structures of 8, 9 and 10 whereby the steric shielding increases in the order Boc < Z < ZAla (see Fig. 1). In the same order the activity decreases. (2) Adding PPh3 to these complexes blocks a coordination site, further increases the steric shielding and makes olefin binding difficult. Low catalytic activity is the result. (3) When the chloro ligand in 8 is exchanged for PF6− and an ion pair like 12 is generated, the slower oxidative H2 may be the reason for the lower catalytic activity. (4) In reactions with [Rh(cod)(ZSerPhos)](PF6), addition of PPh3 likely leads to a “classical” solvated diphosphane complex fragment [Rh(PPh3)(RSerPhos)]+ in which only the phosphorus atom of the SerPhos ligand binds to Rh. This complex then shows the usual high catalytic activity for cationic rhodium bis(phosphane) complexes.2
Conclusions
In rhodium chloro complexes with phosphanyl- or arsanyl-substituted serine derivatives, SerPhos or SerArs, an intramolecular (N)H⋯Cl(Rh)-hydrogen bridge is observed in the solid state and in solution. As a result, the amino acid residue adopts a α-helix type structure. Removal of the chloro ligand from the coordination sphere and consequently cleavage of the NH–Cl bridge, leads to a structural change of the amino acid residue to a β-sheet type. This structural transformation which is triggered by a change in the coordination sphere of the transition metal is fully reversible.In contrast to established rhodium hydrogenation catalysts, the neutral chloro [RhCl(cod)(SerPhos)] complexes give rise to significantly more active catalysts than the corresponding cationic complexes [Rh(cod)(κ2-ZSerPhos)]+. Electronic and, especially, steric reasons may be responsible for this observation. These results encourage to use also larger mono-phosphanyl substituted peptides as ligands for catalysts instead of the formerly investigated disubstituted ones.
In view of the enormous potential of proteins as ligands for catalytically active-transition metal complexes, we also hope that the presented structural and spectroscopic data obtained with the small models discussed in this paper may serve for the design and better understanding of the interaction of metal complex fragments with peptides.
Experimental
General techniques
All syntheses were performed in flame-dried glassware under an atmosphere of argon using standard Schlenk techniques. Solvents were freshly distilled from sodium/benzophenone (thf), from sodium/tetraglyme/benzophenone (hexane, toluene) or calcium hydride (dichloromethane) prior to use. Air sensitive compounds were stored and weighed in an argon filled glovebox (Braun MB 150 B–G system) and reactions on small scale were performed directly in the glovebox.NMR spectra were either taken on an AMX-500, Avance DRX-400, Avance DPX-300, or Avance DPX-250 system. The chemical shifts are given as dimensionless δ values. Spectra were referenced with external standards: for 1H and 13C NMR with TMS, for 15N NMR with NH3, for 19F NMR with CFCl3, for 31P NMR with H3PO4 and for 103Rh NMR with the frequency reference Ξ = 3.16 MHz. Coupling constants J are given in Hertz [Hz] as positive values regardless of their absolute signs. The multiplicity of the signals is indicated as s, d, t, q or m for singlets, doublets, triplets, quartets or multiplets, respectively. Quaternary carbons are indicated as Cquat, aromatic as Car, when not noted otherwise. IR-spectra were measured on a Perkin-Elmer 2000 FT-IR spectrometer using a KBr beamsplitter. The absorption bands are described as follows: very strong (ss), strong (s), middle (m), weak (w), or broad (br). The UV/Vis-spectra were measured with the UV-Vis Lambda 19 spectrometer in 0.5 cm-quartz cuvettes. Melting points were determined with an Büchi melting point apparatus and are not corrected.
O) amide I], 1732.0 (ss, CO), 2941 (m, CH str.), 3324 (ss, NH str.).
[Cu(AsPh2)] 8 : 0.296 g (2.2 mmol) copper(I) tert-butanolate was dissolved in 60 mL THF and cooled to −40 °C. Under vigorous stirring, a solution of 0.5 g (2.2 mmol) diphenylarsane, Ph2AsH, in 10 mL THF was added. After the addition was complete, the deeply coloured solution was stirred for 45 min. at T = −40 °C and subsequently warmed to room temp. Thereby a brown precipitate was formed which was filtered off and washed twice with 10 mL of pentane. Yield: 0.62 g (98%); mp (decomp.): 148 °C. IR (KBr pellet) [ν/cm−1]: 471, 690, 728 (s), 800, 1019 (br), 1260 (m), 1428 (s), 1472 (s).
In the second part of the synthesis, 0.2 mmol copper pnictogenide [Cu(EPh2)]8 (E = P, As) were suspended in 40 mL thf and were vigorously stirred at room temp. Slowly an equimolar amount of the N∩N ligand in 10 mL thf was added and the mixture was stirred for another hour at room temp. The intensely green coloured solutions were concentrated to a few milliliters whereby the products precipitate as fine crystalline powders. These were filtered off, eventually re-crystallized from toluene, and dried under high vacuum. 2: Yield: 53%; mp (decomp.): 135 °C. 1H NMR (C6D6): δ 8.1–6.8 (m, Hligand, Hphenyl), 2.7–2.46 (br, 6H, CH3). 31P NMR (C6D6): −24.0 (br). All 13C-resonances in the aromatic region were strongly broadened. IR (KBr pellet) [ν/cm−1]: 475, 546, 694 (s), 727 (s), 845, 1040.0 (s), 1429.0 (s), 1469.0 (m), 1496 (m), 1574.0 (s), 3040–2860 (m). UV/VIS (toluene) λmax/nm (ε/l mol−1 cm−1): 556 (3192). 6: Yield: 71%; mp (decomp.): 127 °C. 1H NMR (C6D6): δ 7.78 (br, 1H, Hligand), 7.57 (br, 1H, Hligand), 7.32 (br, 1H, Hligand), 7.25 (s, 3H, Hphenyl), 6.94 (br, 3H, Hphenyl), 2.83 (br, 3H, CH3). All 13C-resonances in the aromatic region were strongly broadened. IR (KBr pellet) [ν/cm−1]: 3039 (s, CH str.), 1614, 1569, 1494, 1471, 1427 (s), 1060, 1020, 846 (ss), 728, 695. UV/VIS (thf) λmax/nm (ε/l mol−1 cm−1): 714.6 (217.4).
R(S)-3-Diphenylphosphanylmethyl-N-carbobenzyloxyserinate (ZSerphos) (3a). Yield: 63.3%; mp 68 °C. 1H NMR (C6D6): δ 7.48–7.26 (m, 10H, Haromat), 7.2 (br, 5H, Haromat), 5.48 (br, 1H, N–H), 5.1 (s, 2H, OH2), 4.83 (br, 1H, CH), 3.19 (s, 3H, OCH3), 2.65 (dd, 1H, 2JHCH = 14.1 Hz, 3JHCCH = 5.4 Hz, PH2, 2.41 (dd, 1H, 2JHCH = 14.1 Hz, 3JHCCH = 5.4 Hz, PCH2). 13C NMR (C6D6): δ 171.4 (d, 3JCP = 5.8 Hz, COOCH3), 155.1 (s, Curethan), 136.6 (s, Cipso), 133.8 (d, 1JPC = 17.0 Hz, Cipso), 132.7 (d, 2JPC = 19.8 Hz, Cortho), 128.5 (Cortho), 128.3 (Cpara), 128.3 (d, 3JPC = 14 Hz, Cmeta), 128.1 (Cmeta, Cpara), 66.4 (OCH2), 51.7 (d, 2JCP = 17.4 Hz, CH) 51.2 (s, CO2CH3), 31.8 d, 1JPC = 16.6 Hz, CH2P). 31P NMR (C6D6): δ −23.4. IR (KBr pellet) [ν/cm−1]: 696 (s, monosub. arene), 741 (s, monosub. arene), 1036, 1176 (s, CO), 1205 (m, P–CH2), 1222 (m, P–CH2), 1431 (m, P–C str.), 1502.0 (ss, amide II), 1711 (ss, amide I), 1742.0 (ss, C
O), 2960 (w, CH str.), 3412 (ss, NH str.). Optical rotation [α]20D 0.3 (c 0.01, DMF). Anal. Calc. for C24H24NO4P (421.42 g mol−1) C: 68.4%, H: 5.74, N: 3.32. Found: C: 68.3%, H: 5.74%, N: 3.28%
R(S)-3-Diphenylphosphanylmethyl-N-(tert-butoxycarbonyl)serinate (BocSerphos) (3b). Yield: 53%; Oil. 1H NMR (C6D6): δ 7.47–7.42 (m, 4H, Haryl), 7.13–7.07 (m, 6H, Haryl), 5.77 (br, 1H, NH), 4.81 (m, 1H, CH), 3.24 (s, 3H, CH3), 2.7 (m, 2H, PCH2), 1.42 (s, 9H, C(CH3)3). 13C NMR (C6D6): δ 171.9 (d, 3JCP = 7.3 Hz, C
O), 154.7 (s, Curethan), 133.6 (d, 1JPC = 14.6 Hz, Cipso), 132.7 (d, 2JPC = 17.9 Hz, Cortho), 128.5 (Cpara), 128.3 (d, 3JPC = 14 Hz, Cmeta), 78.7 (s, C(CH3)3), 51.5 (d, 2JPC = 14.9 Hz, CH), 51.1 (s, OCH3), 31.5 (d, 1JPC = 10.05 Hz, PCH2), 27.8 (s, C(CH3)3). 31P NMR (C6D6): δ
−22.8 (s). Anal. Calc. for C21H26NO4P (387.41 g mol−1): C: 65.10%, H: 6.76%, N: 3.61%. Found: C: 65.08%, H: 6.63%, N: 3.63%.
R(S)-3-Diphenylphosphanylmethyl-N-(carbomethoxy)serinate (MetSerphos) (3c). Yield: 67%; oil. 1H NMR (C6D6): δ 7.47–7.42 (m, 4H, Haryl), 7.13–7.07 (m, 6H, Haryl), 5.34 (d, 1H, 3J = 7.7, NH), 4.75 (m, 1H, CH), 3.44 (s, 3H, CH3), 3.19 (s, 3H, CH3), 2.64 (dd, 1H, 2J = 14.0 Hz, 3J = 5.6 Hz, PCH2), 2.38 (dd, 1H, 2J = 13.6 Hz, 3J = 8.0 Hz, PCH2). 13C NMR (C6D6): δ 169.3 (d, 3JPC = 6.5 Hz, COOCH3), 154.4 (s, Curethan), 133.6 (d, 1JPC = 13.6 Hz, Cipso), 132.7 (d, 2JPC = 17.9 Hz, Cortho), 128.5 (d, 3JPC = 7 Hz, Cmeta), 128.1 (Cpara), 53.1 (CO2CH3), 52.9 ppm (COOCH3), 51.1 (s, CH), 32.4 (d, 1JPC = 14.3 Hz, CH2P). 31P NMR (C6D6): δ −24.4 (s). ZAlaSerphos (3d): Yield: 65%; oil. 1H NMR (C6D6): δ 7.64–7.11 (m, 15H, Haryl), 6.1 (br, 1H, NHala), 5.8 (br, 1H, NHser), 5.17 (s, 2H, CH2Ph), 4.99 (m, 1H, CHSer), 4.2 (m, 1H, CHAla), 3.22 (s, 3H, CH3Ser), 2.77 (dd, 1H, 2J = 12.7 Hz, 3J = 5.7 Hz, CH2P), 2.56 (dd, 1H, 2J = 13.8 Hz, 3J = 6.8 Hz, CH2P), 1.21 (d, 3H, 3J = 6.9 Hz, CH3ala). 13C NMR (C6D6): δ 171.5 (d, 3J = 7.5 Hz, COOCH3), 156.5 (s, CONH), 156.1 (s, CONH), 136.7 (s, Cipso), 134.6 (d, 1J = 12 Hz, Cipso), 132.3 (d, 3J = 17.4 Hz, Cortho), 128.6–128.4 (s, Cortho, Cmeta, Cpara, Cmeta, Cpara), 66.4 (s, CH2O) 51.2 (s, CH3O), 50.5 (s, CHala), 50.25 (d, 2J = 11.0 Hz, CHser), 31.7 (d, 1JPC = 17.4 Hz, CH2P), 18.0 (s, CH3ala). 31P NMR (C6D6): δ − 22.5. Anal. Calc. for C27H29N2O5P (492.51 g mol−1): C: 66.85%, H: 5.93%, N: 5.69%. Found: C: 66.81%, H: 6.01%, N: 5.58%.
R(S)-3-Diphenylarsanylmethyl-N-carbobenzyloxyserinate (ZSerars) (7). Yield: 62%; oil. 1H NMR (C6D6): δ 7.47–7.29 (m, 4H, Haromat), 7.19–7.09 (m, 11H, Haromat), 5.51 (d, 1H, 3J = 8.1 Hz, NH), 5.11 (s, 2H, OCH2), 4.81 (br, 1H, CH), 3.17 (s, 3H, OCH3), 2.60 (dd, 1H, 2JHCH = 12.9 Hz, 3JHCCH = 5.7 Hz, PCH2), 2.33 (dd, 1H, 2JHCH = 14.1 Hz, 3JHCCH = 5.4 Hz, PCH2). 13C NMR (C6D6): δ 171.8 (s, COOCH3), 155.2 (s, Curethan), 139.7 (s, Cipso), 136.6 (s, Cipso), 132.8 (s, Cortho), 132.8 (s, Cortho), 128.7 (Cmeta), 128.5 (Cmeta), 128.3 (s, Cpara), 128.2 (s, Cpara), 66.5 (OCH2), 51.7 (s, CH), 51.2 (s, CO2CH3), 31.2 (s, CH2As).
Oester), 168.37 (s, COsuccin), 155.03 (s, Curethan), 135.7 (s, Cipso), 128.25, 127.96, 127.9 (Cortho, Cmeta, Cpara), 66.98 (s, CH2O), 47.8 (s, CH), 25.25 (s, CH2succin), 18.30 (s, CH3).
Chloro(η4-1,5-cyclooctadiene)[R(S)-3-diphenylphosphanylmethyl-N-(carbobenzyloxy)serinate]rhodium(I) [RhCl(ZSerphos)(cod)] (8). Yield: 93%; mp 138 °C. 1H NMR (CD3CN): δ 7.93 (m, 2H, Haryl), 7.51–7.34 (m, 13H, Haryl), 7.18 (d, 1H, 3J = 7.45 Hz, NH), 5.41 (br, 4H, –CH
), 5.17 (m, 2H, OCH2), 4.70 (m, 1H, CH), 3.72 (s, 3H, OCH3), 2.98–2.90 (m, 2H, PCH2), 2.26 (m, –CH2–), 2.02 (m, –CH2–). 13C NMR (CD3CN): δ 171.9 (s, COOCH3), 155.8 (s, CONH), 136.8 (s, Cipso), 134.5 (d, 1JPC = 12.1 Hz, Cipso) 132.1 (d, 4JPC = 9.6 Hz, Cortho), 130.8 (s, Cortho), 130.1 (s, Cmeta), 128.4 (s, Cpara), 128.6 (s, Cpara), 127.9 (d, 3JPC = 6.5 Hz, Cmeta), 71.2 (br, CHCH), 66.4 (s, CH2O), 52.1 (s, CH3), 51.6 (s, CH), 32.0 (br, CH2 (cod)), 28.3 (d, 1JPC = 26.2 Hz, PCH2). 31P NMR (CD3CN): δ 20.6 (d, 1JRhP = 151.7 Hz). IR (KBr pellet) [ν/cm−1]: 3307 (s, NH str.), 2915, 2877 (s, CH str.), 1741 (s, CO), 1707 (ss, (CO) amide I), 1507 (ss, (N–H) amide II), 1430 (m), 1381, 1354, 1260, 1212, (ss), 1041 (s), 1028, 797, 757, 699. UV/VIS (thf) λmax/nm (ε/l mol−1 cm−1): 402.2 (2174), 279.1 (5042). Anal. Calc. for C32H36ClNO4PRh (667.96 g mol−1): C: 57.54%, H: 5.43%, N: 2.09%, P: 4.64. Found: C: 57.3%, H: 5.4%, N: 2.3%, P: 4.6%.
Chloro(η4-1,5-cyclooctadiene)[R(S)-3-diphenylphosphanylmethyl-N-(tert-butoxycarbonyl)serinate]rhodium(I) [RhCl(BocSerphos)(cod)] (9). Yield: 62%; mp 105 °C. 1H NMR (CDCl3): δ 7.88 (m, 2H, Haryl), 7.53–7.26 (m, 8H, Haryl), 6.82 (d, 1H, 3J = 7.14 Hz, NH), 5.54 (br, 2H, –CH
), 4.72 (m, 1H, CH), 4.24 (br, 2H, CH), 3.73 (s, 3H, OCH3), 3.18–3.03 (m, 2H, PCH2), 2.43 (m, –CH2–), 2.04 (m, –CH2–), 1.52 (s, 9H, C(CH3)3). 13C NMR (CDCl3): δ 172.1 (d, 3JPC = 13.1 Hz, COOCH3), 155.5 (s, CONH), 134.5 (d, 1JPC = 11.8 Hz, Cipso) 132.1 (d, 4JPC = 9.7 Hz, Cortho), 129.6 (s, Cpara), 128.2 (d, 3JPC = 8.2 Hz, Cmeta), 104.5 (m, CH), 78.4 (s, C(CH3)3), 70.5 (dd, 1JRhC = 53.7 Hz, 2JCP = 13.7 Hz, CHCH), 66.4 (s, CH2O), 52.2 (s, CH3), 51.1 (s, CH), 32.9, 32.2, 30.5 (s, CH2 (cod)), 29.1 (d, 1JPC = 26.1 Hz, PCH2), 28.6 (s, CH2 (cod)), 28.14 (s, C(CH3)3). -31P NMR (CDCl3): δ 20.8 (d, 1JRhP = 150.0 Hz). IR (KBr pellet) [ν/cm−1]: 3296 (s, NH str.), 2962–2830 (s, CH str.), 1754 (ss, CO), 1697 [ss, (CO) amide I], 1504 [ss, N–H amide II], 1431 (m, P–C str.), 1362, 1258 (m, P–CH2), 1214, (m), 1160 (s, C–O), 1096 (s), 1017, 801 (s, monosub. arene), 744 (s, monosub. arene), 691. UV/VIS (thf) λmax/nm (ε/l mol−1 cm−1): 400.1 (1681), 287.1 (5500). Anal. Calc. for C29H38ClNO4PRh (633.95 g mol−1): C: 54.94%, H: 6.04%, N: 2.21%. Found: C: 54.3%, H: 5.97%, N: 2.3%.
Chloro(η4-1,5-cyclooctadiene)[R(S)-3-diphenylarsanylmethyl-N-(carbobenzyloxy)serinate]rhodium(I) [RhCl(ZSerars)(cod)] (11). Yield: 35.5%; mp 126 °C. 1H NMR (CD3CN): δ 7.70–7.66 (m, 2H, Haryl), 7.49–7.2 (m, 13H, Haryl), 6.98 (d, 1H, 3J = 7.62 Hz, NH), 5.11 (dd, 2H, 2J = 16.3 Hz, OCH2), 4.59 (m, 1H, CH), 4.27 (s, 4H, –CH
), 3.68 (s, 3H, OCH3), 2.89–2.77 (m, 2H, AsCH2), 2.7 (m, –CH2–), 1.86–1.79 (m, –CH2–). 13C NMR (CD3CN): δ 172.4 (s, COOCH3), 156.2 (s, CONH), 137.4 (s, Cipso), 134.3 (s, Cipso) 130.6 (s, Cortho), 130.35 (s, Cpara), 129.4 (s, Cmeta), 128.9 (s, Cortho), 128.5 (s, Cpara), 128.3 (s, Cmeta), 82.2 (br, CHCH), 66.8 (s, CH2O), 52.6 (s, CH3), 52.1 (s, CH), 31.0 (s, CH2 (cod)), 28.0 (s, AsCH2). IR (KBr pellet) [ν/cm−1]: 3272 (s, NH str.), 2911, 2872 (s, CH str.), 1743 (ss, CO), 1710 (s, amide I, CO), 1527 (s, amide II), 1431 (m), 1211 (ss), 1044, 1025, 734, 693. UV/VIS (thf) λmax/nm (ε/l mol−1 cm−1): 374.8 (1242), 277.1 (5000). Anal. Calc. for C32H36AsClNO4Rh (711.91 g mol−1): C: 53.99%, H: 5.10%, N: 1.97%. Found: C: 47.5%, H: 5.14%, N: 1.3%.
), 5.17/5.13 (s, 2H, OCH2Ph), 4.84/4.70 (dddd, 3Jα,β = 2.8/3.0 Hz, 3Jα,β′ = 13.0/12.0 Hz, 3Jα,NH = 6.7/6.8 Hz, 3Jα,P = —/8.8 Hz, 1H, CHSer), 4.60/4.09 (m, 1H, CHAla), 3.72/3.69 (s, 3H, OCH3), 3.27/3.01 (ddd, 2Jβ,P = 6.6/3.8 Hz, 3Jα,β = 12.0/13.0 Hz, 2Jβ,β′ = 14.7 Hz/—, 1H, CH2P), 2.87/2.85 01 (ddd, 2Jβ′,P = 12.2/14.2 Hz, 3Jα,β′ = 3.0/2.8 Hz, 2Jβ,β′ = 14.7 Hz/—, 1H, CH2P), 2.45/2.09 (br, 4H, –CH2– (cod)), 2.34/1.90 (br, 4H, –CH2– (cod)), 1.49/1.44 (d, 3JH,H = 7.0/7.0 Hz, CH3Ala). 13C NMR (CD2Cl2): δ 173.0/172.5 (s, COOCH3), 171.9/171.7 (s, CONH), 156.1/155.9 (s, CONH), 137.3/137.3 (s, Cipso), 135.4/134.8 (d, 1JP,C = 12.3/12.3 Hz, Cipso), 133.1/132.6 (d, 2JP,C = 9.9/9.9 Hz, Cortho), 128.6/128.2 (d, 3JP,C = 7.0/7.0 Hz, Cmeta), 131.5/131.1 (s, Cortho), 130.7/130.7 (s, Cmeta), 128.9/128.8 (s, Cpara), 128.7/128.7 (s, Cpara),73.3/72.2/71.5 (d, J = 13.5 Hz, CH (cod)), 66.9/66.9 (s, CH2Ph), 58.5/58.5 (s, OCH3), 51.0/51.0 (s, CHAla), 50.7/50.4 (s, CHSer), 33.5/33.2/32.7 (br, CH2 (cod)), 29.1/28.7 (d, 1JC,P = 23.2/23.2 Hz, CH2P), 20.1/19.7 (s, CH3(Ala)). 31P NMR (CD2Cl2) δ 20.3/18.4 (d, 1JP,Rh = 150.7/150.7 Hz). 103Rh NMR (CD2Cl2) δ 372/365. IR (KBr pellet) [ν/cm−1]: 3258 (s, NH str.), 1735 (ss, CO), 1717 [ss, (CO) amide I], 1509 [ss, (N–H) amide II]. Anal. Calc. for C35H41ClN2O5PRh (739.05 g mol−1): C: 56.88%, H: 5.59%, N: 3.79%. Found: C: 56.90%, H: 5.61%, N: 3.78%.
), 5.01 (m, 2H, OCH2), 4.84 (m, 1H, CH), 4.33 (br, 2H, –CH), 3.85 (s, 3H, OCH3), 3.23–2.80 (m, 2H, PCH2), 2.47 (m, –CH2–), 2.08 (m, –CH2–). 13C NMR (CDCl3): δ 171.9 (s, COOCH3), 158.5 (s, CONH), 136.3 (d, 1JPC = 10.1 Hz, Cipso), 134.5 (s, Cipso), 133.7 (d, 4JPC = 12.4 Hz, Cortho), 131.8 (s, Cortho), 131.7 (s, Cmeta), 130.98 (s, Cpara), 129.1 (s, Cpara), 128.8 (d, 3JPC = 6.8 Hz, Cmeta), 108.5 (m, CHCH), 71.3 (br, CHCH), 68.3 (s, CH2O), 53.9 (s, CH3), 52.55 (s, CH), 31.9 (br, CH2 (cod)), 27.5 (d, 1JPC = 26.8 Hz, PCH2). 31P NMR (CDCl3): δ 25.7 (d, 1JPRh = 148.5 Hz, RhPCH2), −143.0 (sept, 1JPF = 713 Hz, PF6−). IR (KBr pellet) [ν/cm−1]: 3384 (br, NH str.), 2918, 2876 (s, CH str.), 1725 (s, CO), 1623 (ss, amide I), 1432 (m), 1382, 1259, 1218, 1096 (s), 1026, 839 (ss, PF str.), 695. Anal. Calc. for C29H38F6NO4P2Rh (777.48): C: 49.43%, H: 4.67%, N: 1.8%. Found: C: 50.4%, H: 4.9%, N: 2.0%.
), 5.10/5.08 (s, 2H, OCH2Ph), 5.14/4.90 (d, 6.6/5.9 Hz, 1H, NHAla), 4.04/3.75 (m, 1H, CHAla), 4.00/3.97 (s, 3H, OCH3), 3.41/3.35 (br, 2H, –CH
trans to P), 3.21/3.16 (br, 2H, –CH
trans to O), 3.05/2.95 (m, 2H, CH2P), 2.70–1.90 (br, 8H, CH2 (cod)), 0.82/0.75 (d, 3J = 7.3/7.0 Hz, 3H, CH3Ala). 13C NMR (CD2Cl2): δ 179.4/179.3 (br, CAlaONH), 169.3/169.1 (d, 3JCP = 17.2/18.3 Hz, CSerONH), 156.3/156.3 (s, COcbz), 136.1/136.1 (s, Cipso), 134.7/134.4 (d, 2JPC = 13.0/13.3 Hz, Cortho), 129.6/129.4 (d, 1JPC = 11.1/7.5 Hz, Cipso), 136–126 (Caryl), 110.1/109.5 (m (br), CH (cod) trans to P), 71.0/69.9 (m (br), CH (cod) trans to O), 67.3/67.2 (s, OCH2Ph), 54.3/54.3 (s, OCH3), 52.9/52.9 (s, CHSer), 51.8/51.2 (s, CHAla), 35.0/35.4 (m, CH2P), 34.0/32.1/28.7/27.1 (CH2 (cod)), 15.9/15.9 (s, CH3(Ala)). 31P NMR (CD2Cl2): δ 27.9/27.9 (d, 1JPRh = 151.8 Hz, PPh2), −144.4 (sept, 1JPF = 712 Hz, PF6−). 103Rh NMR (CD2Cl2): δ 426/413. IR [ν/cm−1]: 3330 (s, NH str.), 1723 (ss, CO), 1613 (ss, (CO) amide I). Anal. Calc. for C35H41F6N2O5P2Rh (848.55 g mol−1): C: 49.54%, H: 4.87%, N: 3.30%. Found: C: 49.67%, H: 4.88%, N: 3.29%.
), 5.21 (m, 2H, OCH2), 4.77 (m, 1H, CH), 4.23 (br, 2H, –CH), 3.75 (s, 3H, OCH3), 3.17–2.88 (m, 2H, PCH2), 2.48–1.75 (m, 8H, –CH2–), 2.01 (s, 3H, CH3CN). 13C NMR (CDCl3): δ 171.8 (d, 3JPC = 13.1 Hz, COOCH3), 156.5 (s, ONH), 136.05 (s, Cipso), 134.4 (d, 1JPC = 11.6 Hz, Cipso), 132.0 (d, 4JPC = 9.44 Hz, Cortho), 130.6 (s, Cortho), 129.9 (s, Cmeta), 128.3 (d, 3JPC = 5.4 Hz, Cmeta), 128.1 (s, Cpara),127.6 (s, Cpara), 117 (N), 104.9 (m, CHCH), 71.0 (br, CHCH), 66.7 (s, CH2O), 52.5 (s, CH3), 51.34 (s, CH), 32.8, 32.1, 30.56 (br, CH2 (cod)), 29.1 (d, 1JPC = 26.5 Hz, CPH2), 28.27 (br, CH2 (cod)) 1.73 (s, CH3CN). 31P NMR (CDCl3): δ 20.4 (d, 1JPRh = 149.4 Hz, RhPCH2), −143.0 (sept, 1JPF = 711 Hz, PF6−). IR (KBr pellet) [ν/cm−1]: 3400 (br, NH str.), 2951, 2877 (s, CH str.), 1727 (s, CO), 1708 (ss, amide I), 1503 (s, amide II), 1430 (m), 1381, 1258, 1094 (s), 1027, 800 (s, PF str), 695.
X-Ray crystallography (see Table 6)
Single crystals were selected in an argon filled glovebox. The data were collected on a Picker four-circle, Stoe upgraded, diffractometer (8, 9) and a Siemens CCD (10, 11) diffractometer. For 8 and 9 an experimental absorption correction (integration from crystal shape) was performed. For 10 and 11, an absorption correction was applied using the program SADABS. The structures were solved by direct methods, all non-hydrogen atoms were refined against F2 (G. M. Sheldrick, SHELXL-97, Göttingen, 1993, 1997) with anisotropic temperature factors while the hydrogen atoms were constrained using a riding model.| 8 | 9 | 10 | 11 | |
|---|---|---|---|---|
| Formula | C32H36ClNO4PRh | C29H38ClN1O4PRh·0.5CH3CN | C35H40ClN2O5PRh | C32H36AsClNO4Rh |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group | P21/c | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| Z | 8 | 4 | 2 | 1 |
| T/K | 293(2) | 293(2) | 293(2) | 293(2) |
| D c/g cm−3 | 1.428 | 1.391 | 1.301 | 1.511 |
| a/Å | 21.339(2) | 15.575(8) | 10.7543(3) | 8.3011(2) |
| b/Å | 30.582(2) | 10.255(5) | 11.7575(2) | 9.7422(3) |
| c/Å | 9.520(5) | 19.57(1) | 16.8367(4) | 10.7146(3) |
| α/° | 90 | 90 | 104.312(1) | 87.201(1) |
| β/° | 90.55(5) | 90.06(4) | 91.873(1) | 76.277(1) |
| γ/° | 90 | 90 | 112.649(1) | 68.507(1) |
| V/Å3 | 6212(7) | 3126(3) | 1884.17(8) | 782.54(4) |
| µ/mm−1 | 6.015 | 5.965 | 0.606 | 1.716 |
| Crystal size/mm | 0.2 × 0.1× 0.1 | 0.2 × 0.15 × 0.1 | 0.2 × 0.1 × 0.1 | 1.0 × 0.6 × 0.4 |
| Radiation (λ/Å) | Cu-Kα; graphite monochromator (1.54178) | Mo-Kα; graphite monochromator (0.71073) | ||
| 2θ range/° | 4.14 ≤ 2θ ≤ 85.00 | 5.68 ≤ 2θ ≤ 99.96 | 2.52 ≤ 2θ ≤ 56.56 | 3.92 ≤ 2θ ≤ 65.40 |
| Index ranges, hkl | −18 to 18, 0 to 26, 0 to 8 | −15 to 11, 0 to 10, 0 to 19 | −14 to 13, −12 to 15, −22 to 17 | −11 to 12, −14 to 13, −15 to 8 |
| No. of rflns total | 4357 | 2998 | 15731 | 8075 |
| No. of unique rflns | 4357 | 2998 | 9127 | 6090 |
| No. of parameters/restraints | 649/0 | 356/0 | 433/0 | 361/3 |
| R 1 (rflns I > 2σ(I)) | 0.0440 | 0.0445 | 0.0557 | 0.0423 |
| wR 2 (all data) | 0.1216 | 0.1193 | 0.1714 | 0.1076 |
| Max., min. residual electron density/e Å−3 | 0.563, −0.406 | 1.002, −0.553 | 1.301, −0.766 | 1.068, −1.015 |
The asymmetric unit of 9 contains half a molecule of acetonitrile, disordered over two positions which were refined with an occupancy factor of 0.5 for each site. Two diastereotopic molecules were found in the elementary cell of 10. While the racemic SerPhos residue fulfils the symmetry properties of the centrosymmetric space group, this is not the case for the alanine. Hence the methyl group of the alanine was found on two different positions and was refined as a disordered group with an occupancy factor of 0.5 for each site.
CCDC reference numbers 178294, 178295, 283233 and 283234.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512653c
References
- A. Agarkov, S. J. Greenfield, T. Ohishi, S. E. Collibee and S. R. Gilbertson, J. Org. Chem., 2004, 69, 8077 CrossRef CAS . For a review, see: S. R. Gilbertson, Prog. Inorg. Chem., 2001, 50, 433 Search PubMed.
- See, for example: Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2002 Search PubMed.
- For experimentally determined binding constants, see: (a) C. M. Haar, S. P. Nolan, W. J. Marshall, K. G. Molloy, A. Prock and W. P. Giering, Organometallics, 1999, 18, 474 CrossRef CAS; (b) D. C. Smith, Jr., C. M. Haar, E. D. Stevens and S. P. Nolan, Organometallics, 2000, 19, 1427 CrossRef.
- See, for example: (a) H. Park and T. V. RajanBabu, J. Am. Chem. Soc., 2001, 124, 734 , and references therein; (b) T. V. RajanBabu, T. A. Ayers and A. L. Casalnuovo, J. Am. Chem. Soc., 1994, 116, 4101 CrossRef CAS; (c) A. Kumar, G. Oehme, J. P. Roque, M. Schwarze and R. Selke, Angew. Chem., 1994, 106, 2272 CrossRef; A. Kumar, G. Oehme, J. P. Roque, M. Schwarze and R. Selke, Angew. Chem., Int. Ed. Engl., 1994, 33, 2197 CrossRef; (d) R. Selke, J. Organomet. Chem., 1989, 370, 249 CrossRef CAS; (e) R. Selke, J. Organomet. Chem., 1989, 370, 241 CrossRef CAS; (f) V. Šunjić, I. Habuš and G. Snatzke, J. Organomet. Chem., 1989, 370, 295 CrossRef CAS; (g) V. A. Pavlov, E. I. Klabunovskii, Y. T. Struchkov, A. A. Voloboev and A. I. Yanovsky, J. Mol. Catal., 1988, 44, 217 CrossRef CAS.
- See, for example: S. R. Gilbertson, S. E. Collibee and A. Agarkov, J. Am. Chem. Soc., 2000, 122, 6522 Search PubMed , and references therein.
- G. Xu and S. R. Gilbertson, Tetrahedron Lett., 2002, 43, 2811 CrossRef CAS.
- (a) S. R. Gilbertson and X. Wang, Tetrahedron, 1999, 55, 11609 CrossRef CAS; (b) S. R. Gilbertson and X. Wang, Tetrahedron Lett., 1996, 37, 6475 CrossRef CAS.
- (a) I. Grassert, K. Schinkowski, D. Vollhardt and G. Oehme, Chirality, 1998, 10, 754 CrossRef CAS; (b) I. Grassert, V. Vill and G. Oehme, J. Mol. Catal. A, Chem., 1997, 116, 231 CrossRef CAS.
- S. R. Gilbertson, G. Chen, J. Kao, A. Beatty and C. F. Campana, J. Org. Chem., 1997, 62, 5557 CrossRef CAS.
- (a) C. Meyer, H. Grützmacher and H. Pritzkow, Angew. Chem., 1997, 109, 2576 CrossRef; C. Meyer, H. Grützmacher and H. Pritzkow, Angew. Chem., Int. Ed. Engl., 1997, 36, 2471 CrossRef CAS; (b) H. Grützmacher, C. Meyer, S. Boulmaâz, H. Schönberg, S. Deblon, J. Liedtke, S. Loss and M. Wörle, Phosphorus Sulfur Silicon, 1999, 114, 465 Search PubMed.
- D. J. Brauer, K. W. Kottsieger, S. Schenk and O. Stelzer, Z. Anorg. Allg. Chem., 2001, 627, 1151 CrossRef CAS.
- S. J. Greenfield and S. R. Gilbertson, Synthesis, 2001, 15, 2337.
- See, for example: (a) H. J. Dyson and P. Wright, Annu. Rev. Biophys. Chem., 1991, 20, 519 CrossRef CAS; (b) M. Williamson and J. P. Waltho, Chem. Soc. Rev., 1992, 227 RSC; (c) C. Altona, R. Francke, R. De Haen, J. Ippel, G. Daalmans, A. Hoekzema and J. Van Wijk, Magn. Reson. Chem., 1994, 32, 670 CAS; (d) A. C. Wang and A. Bax, J. Am. Chem. Soc., 1996, 118, 2483 CrossRef CAS , and references therein.
- (a) K. Wüthrich, NMR of Proteins and Nucleic Acids, Wiley, New York, 1986 Search PubMed; (b) V. Bystrov, Prog. Nucl. Magn. Reson. Spectrosc., 1976, 10, 41 CrossRef.
- Shifts by 50–100 cm−1 for ν(CO) to lower wavenumbers and downfield (high frequency) shifts by ≈7 ppm of the 13C resonance of the coordinating amide group in P,CO ligands were reported:
(a) S. Sjövall, L. Kloo, A. Nikitidis and C. Anderson, Organometallics, 1998, 17, 579 CrossRef;
(b) M. Kuriyama, K. Nagai, K.-i. Yamada, Y. Miwa, T. Taga and K. Tomioka, J. Am. Chem. Soc., 2002, 124, 8932 CrossRef CAS.
- Alternatively one may speculate that the alanine residue is even more fluxional in 10 and the sharper NMR lines stem from averaged signals even at low temperature.
- D. Savithri, C. Leumann and R. Scheffold, Helv. Chim. Acta, 1996, 79, 288 CrossRef CAS.
- M. Monsigny, D. Delay and M. Vaculik, Carbohydr. Res., 1977, 59, 589 CrossRef CAS.
- D. Theodoropoulos, I. Schwartz and R. Walter, Biochemistry, 1967, 6, 3927 CrossRef CAS.
- T. H. Lemmen, G. V. Goeden, J. C. Huffman, R. L. Geerts and K. G. Caulton, Inorg. Chem., 1990, 29, 3680 CrossRef CAS.
- M. Santamaria, T. Maina, U. Mazzi and M. Nicolini, Inorg. Chim. Acta, 1995, 240, 291 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |