DOI:
10.1039/B512074H
(Paper)
Dalton Trans., 2006, 121-128
Mixed hydrazido amido/imido complexes of tantalum, hafnium and zirconium: potential precursors for metal nitride MOCVD
Received
25th August 2005
, Accepted 17th October 2005
First published on 1st November 2005
Abstract
The coordination chemistry of the hydrazine derivatives dimethylhydrazine (Hdmh) and N-trimethylsilyl-N′N′-dimethylhydrazine (Htdmh) at Ta, Zr and Hf was investigated aiming at volatile mixed ligand all-nitrogen coordinated compounds. The hydrazido ligands were introduced either by salt metathesis employing the Li salts of the hydrazines and the tetrachlorides MCl4 (M = Zr, Hf) or by amine substitution using M(NR2)4 (R = Me, Et) and [(t-BuN)Ta(NR2)3]. The new complexes were fully characterised including 1H/13C NMR, mass spectrometry and a study of their thermal behaviour. The crystal structures of [ZrCl(tdmh)3] and the all-nitrogen coordinated complex [Ta(N-t-Bu)(NMe2)2(tdmh)] are discussed as well as the structure of the by-product [Li(tdmh)(py)]2. Preliminary MOCVD experiments of the liquid compound [Ta(NEt2)2(N-t-Bu)(tdmh)] were performed and the deposited TaN(Si) films were analysed by RBS and SEM.
Introduction
Metal–organic complexes of early transition metals, bearing nitrogen containing ligands received increasing attention over the last couple of years. One major application of these compounds is their use as precursors for the chemically robust conducting metal nitride (MN, M = Zr, Hf, Ta) thin films via MOCVD and ALD techniques. These films can serve either as a diffusion barrier for copper in copper–silicon metallisation schemes as well as gate electrode materials to replace polycrystalline silicon.1 Also the non-conducting nitride materials, e.g. Ta3N5-films, bear applications, for example as photoelectrode materials.2 The compounds that can be used for MOCVD and ALD processes have to meet several requirements including volatility, purity, a good decomposition behaviour and thermal stability. Tuning stability and reactivity of the precursors is crucial for ALD-purposes in particular, because these thermal properties set limits for the maximum substrate temperature that can be used during deposition.3 So far in most of the reports about the deposition experiments of TaN-thin films, tantalum pentachloride or well-known and partly commercially available mixed amido/imido-complexes e.g. [Ta(NEt2)3(N-t-Bu)] (TBTDET), [Ta(NMe2)3(NC(CH3)2CH2CH3)] (Taimata), [Ta(NMe2)5] (PDMAT), [Ta(NEt2)5] (PDEAT) and [Ta(NMe2)3(N-t-Bu)] (Et = ethyl) were used.4 Especially Taimata shows a promising behaviour for the deposition of conducting TaN thin films via MOCVD. Other precursors that have been studied for their behaviour in MOCVD are [Ta(NEt2)(NCy2)2], [TaCl3(NSiMe3)(NC5H3Me2-3,5)2] and precursors, based on guanidinate ligand systems.4,5 In the case of zirconium and hafnium, mostly amido or halide complexes were used for deposition experiments.6 Although several experiments seem to be partly successful, oxidation of the nitride thin films and the formation of non-conducting Zr3N4, Hf3N4 and Ta3N5 occurs. This turns out to be a major problem in this area of application.
Ligands derived from hydrazines (Scheme 1) recently gained some attention in the precursor chemistry of metal nitrides. More or less volatile complexes of the type [M1(tdmh)3] (M1 = In, Ga)7 and [M2(MeN–NMe2)4] (M2 = Ti, Zr, Hf)8 were reported and partially tested for MOCVD. Reactions of dimethylhydrazine with TaCl5 and the formation of [Ta(NNMe2)(N(H)NMe2)(N(H2)NMe2)] were reported by Winter et al., but apparently this work has not yet been continued.9 We decided to expand the use of Htdmh as a ligand system on the metals zirconium, hafnium and tantalum based on the following reasoning.
 |
| | Scheme 1 | |
Hydrazine derivatives are known for their reductive capabilities that should support the formation of conducting MN (M = Ti, Zr, Hf, Ta).10
The absence or the reduced number of metal-nitrogen-carbon bonds should reduce the incorporation of carbon into the deposited films.
Incorporation of the silicon from the tdmh ligand into the films could significantly improve the thermal stability and the stability towards oxidation of the thin films.11
The reductive capabilities of dimethylhydrazine allowed the MOCVD of conductive, cubic HfN using [Hf(NEt2)4].12 It can be assumed that intermediate Hf-hydrazido complexes play a significant role in the reduction of the Hf(IV)-centre in this type of separate source MOCVD processes. Therefore, synthesising the “intermediate” mixed amido/imido/hydrazido complexes of Zr, Hf and Ta would be very interesting with respect to their behaviour as MOCVD precursors. Two different approaches were chosen in order to introduce the hydrazine ligand into the complex. One way would be a salt metathesis reaction between the lithiated hydrazine and a metal chloride compound. The other possibility would be a substitution of amido ligands by the hydrazine derivative, by some sort of transamination reaction, at an already fully nitrogen coordinated metal complex. The synthesised new compounds were fully characterised including the determination of the structure in the solid state of selected examples by single-crystal X-ray diffraction. Furthermore, the thermal properties of 3–7 are evaluated by TG/DTA in view of their application in ALD or MOCVD processes. For [Ta(N-t-Bu)(NEt2)2(tdmh)], preliminary MOCVD studies were performed in order to test its suitability as an precursor for the deposition of TaN. These results are one of the few examples of MOCVD-experiments applying hydrazido containing precursors.
Results and discussion
Below, two possible approaches for introducing the tdmh-ligands into Zr, Hf and Ta complexes are presented. Crystal structures are discussed with respect to the bonding behaviour of the ligands in comparison to known amido, imido and hydrazido complexes of the studied metals.
Introduction by salt metathesis
Salt metathesis reactions, applying the lithiated hydrazine ligand seem to be the straight forward way of introducing hydrazido groups into transition metal complexes. These reactions are adequate to the synthesis of most of the early transition metal amido complexes such as [Zr(NR2)4], [Hf(NR2)4] and [Ta(NR2)3(N-t-Bu)] (R = Me, Et) from the metal chlorides and the lithiated amines. Therefore reactions were carried out, using the zirconium and hafnium chlorides and miscellaneous equivalents of the lithiated Htdmh-ligand. An excess of the ligand (3–5 equivalents) resulted in the formation of the complexes [Zr(tdmh)3Cl] and [Hf(tdmh)3Cl] (Scheme 2). It was not possible to derive the homoleptic tetracoordinated congeners [M(tdmh)4]. Presumably steric reasons play a role when the tdmh ligand is introduced instead of trimethylhydrazine.8 The compounds are white solids at room temperature with melting points above 100 °C. Elemental analysis show sufficient purity of the sublimed products. 1H and 13C NMR spectra revealed only two peaks for each compound, which indicates that the three tdmh ligands are chemically equivalent due to fluxional processes in solution. In the case of the zirconium compound, crystals, suitable for single-crystal X-Ray diffraction analysis were obtained from a hexane solution of 1 at −30 °C. The overall molecular geometry of [ZrCl(tdmh)3] with the atomic labelling scheme is shown in Fig. 1. Selected bond lengths and angles are listed in Table 1 and crystallographic data is presented in Table 3. The molecule of 1 comes close to a Cs symmetry where the mirror plane contains the set of atoms [Zr(1)–Cl(1)–N(1)–N(2)–Si(1)]. The structure of [ZrCl(tdmh)3] is similar to that of [TiCl(tdmh)3].8 On the basis of Zr–N distances, [ZrCl(tdmh)3] can be described as having three η2-tdmh ligands. The bond length of the direct bonds (N(2)–Zr(1), N(4)–Zr(1) and N(6)–Zr(1), average: 2.080 Å) are significantly shorter than the donating bonds between Zr and the free electron pair of the nitrogen atoms N(1), N(3) and N(5) (average: 2.295 Å). The bond lengths are close to those observed in other zirconium hydrazido complexes.8 The three trimethylsilyl groups are pointing towards the Cl ligand which is probably due to the larger steric demand of the NMe2-groups in comparison to the NSiMe3 groups. The relative small bite angles of 38° are in accordance to the angles observed in the homoleptic complex [Zr(MeN–NMe2)4].8
Table 1 Selected bond lengths (Å) and angles (°) for the crystal structure of 1
| Zr(1)–N(1) |
2.300(4) |
N(2)–Zr(1)–N(4) |
119.77(13) |
| Zr(1)–N(2) |
2.074(3) |
N(2)–Zr(1)–N(6) |
122.53(13) |
| Zr(1)–N(3) |
2.298(4) |
N(4)–Zr(1)–N(6) |
117.54(12) |
| Zr(1)–N(4) |
2.081(3) |
N(1)–Zr(1)–N(2) |
37.66(13) |
| Zr(1)–N(5) |
2.287(4) |
N(3)–Zr(1)–N(4) |
38.06(12) |
| Zr(1)–N(6) |
2.085(3) |
N(5)–Zr(1)–N(6) |
38.36(12) |
| Zr(1)–Cl(7) |
2.4872(13) |
N(2)–Zr(1)–Cl(1) |
90.38(10) |
| N(1)–N(2) |
1.428(5) |
N(4)–Zr(1)–Cl(1) |
93.51(9) |
| N(3)–N(4) |
1.443(4) |
N(6)–Zr(1)–Cl(1) |
90.18(10) |
| N(5)–N(6) |
1.449(5) |
Zr(1)–N(1)–N(2) |
62.55(17) |
| Si(1)–N(2) |
1.724(3) |
Zr(1)–N(3)–N(4) |
62.78(17) |
| Si(2)–N(4) |
1.725(3) |
Zr(1)–N(5)–N(6) |
63.26(17) |
| Si(3)–N(6) |
1.720(3) |
Zr(1)–N(2)–Si(1) |
146.3(2) |
| |
|
Zr(1)–N(4)–Si(2) |
146.32(18) |
| |
|
Zr(1)–N(6)–Si(3) |
146.6(2) |
| |
|
Zr(1)–N(6)–Si(3) |
146.6(3) |
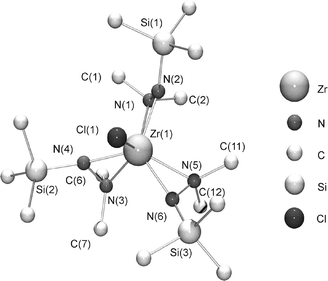 |
| | Fig. 1 Molecular structure of 1 in the solid state. | |
 |
| | Scheme 2 | |
Homoleptic complexes, such as [Ta(tdmh)5] seem not to be reasonable target molecules, when aiming at all-nitrogen coordinated compounds, due to the sterical demand of the hydrazido ligand. Instead we used the well known tantalum complex [TaCl3(N-t-Bu)(py)2] and studied the substitution of the chloro ligands by treatment with different equivalents of the lithiated tdmh ligand. Although indications (1H NMR) were found for the exchange of two chloro ligands of the tantalum compound by two hydrazido ligands, it was not possible to isolate [TaCl(N-t-Bu)(tdmh)2] in significant amounts or sufficient purity. However, we observed the formation of a pyridine stabilized complex of the type [Li(py)(tdmh)]2 (8) as a by-product in these reactions. This complex is described at a later stage in this publication.
Introduction by ligand exchange
As a second approach to introduce the hydrazido ligands, we investigated hydrazine/amine exchange reactions. These transamination type reactions allowed the synthesis of several new zirconium, hafnium and tantalum compounds. The starting materials [Zr(NEt2)4], [Hf(NEt2)4], [Ta(NMe2)3(N-t-Bu)], [Ta(NMeEt)3(N-t-Bu)] and [Ta(NEt2)3(N-t-Bu)] were treated with the ligand Htdmh under various conditions. The steric demand of the hydrazido ligand, typically preferring side-on coordination, significantly limits the number of amido-groups that can be exchanged. In all reactions with tantalum amido/imido complexes and the tetrakis(diethylamido) complexes of zirconium and hafnium, even with a large excess of Htdmh and long periods of refluxing, only one single amido group was replaced (Scheme 3). The assumption that steric hindrance is a limiting factor in the substitution reactions is supported by the increasing reaction time for alkylamido substitution in the order NMe2, NMeEt, NEt2. The reactions were carried out by simply adding a small excess of Htdmh to the slightly yellow solutions of the starting compounds in hexane. Only in the synthesis of 7, a large excess of Htdmh and a refluxing period of 5 days was necessary for quantitative formation of the product. Elemental analysis revealed that the products were synthesised in sufficient purity, with batch sizes up to 50 g.
 |
| | Scheme 3 | |
The crude products show only small amounts of impurities. Nevertheless all compounds were purified by short-path distillation at temperatures between 120 and 143 °C under vacuum conditions (0.01 mbar) prior to further analysis.
NMR and MS analysis of 3–7
1H and 13C NMR spectra were recorded for identification of the compounds. Integration of the peaks clearly shows that one amido group was replaced. As a representative example, the spectroscopic features of compound 6 are discussed here. Due to the chirality of 6, the two diastereotopic protons of the Ta–NCH2CH3 groups are split into two doublets of quartets (Fig. 2). Nevertheless, the two amido-groups are chemically equivalent in solution, resulting in one set of peaks. The methyl groups, bound to one of the hydrazido nitrogen atoms (δ 2.42 ppm) appear as one peak in the NMR spectrum, indicating that rotation along the N–N axis is possible in solution. EI-Mass spectra at 30 or 70 eV provided no valuable information about structural details or decomposition pathways. Only the cleavage of methyl groups can be observed. In all cases, besides 2 and 8, the peaks of the undecomposed molecules appear in the spectra.
![1H NMR of [Ta(NMeEt)2(N-t-Bu)(tdmh)] (6) at room temperature, recorded in C6D6.](/image/article/2006/DT/b512074h/b512074h-f2.gif) |
| | Fig. 2
1H NMR of [Ta(NMeEt)2(N-t-Bu)(tdmh)] (6) at room temperature, recorded in C6D6. | |
Crystal structure of 5
Although 3–7 are viscous liquids at room temperature, it was possible to obtain the molecular structure of 5 by crystallisation at −20 °C from diethyl ether and further preparation and manipulation of the crystals under continuous cooling at −10 °C. The crystals were mounted on a capillary using inert oil and transferred immediately to the cold gas stream (105 K) of the diffractometer. The compound crystallises in the form of colourless prisms. Crystallographic data is presented in Table 3, while selected bond lengths and angles are summarised in Table 2. The molecular structure of 5 in the solid state contains two crystallographic independent molecules in the unit cell. This complex is one of the few examples for all nitrogen five coordinated tantalum complexes (e.g. the homoleptic complex [Ta(NMe2)5]).13 An imaginary mirror plane put through the atoms Ta(1)–N(11)–N(12)–N(13) gives an idea of the basic symmetry of the complex, although no real Cs symmetry exists (Fig. 3). The hydrazido ligand is bound in the expected chelating fashion with a N(11)–Ta(1)–N(12) bite angle of 37.6°. The Ta–N bond lengths to the hydrazido ligand lie in the expected range similar to the alkylamido starting complex and related systems. The Ta(1)–N(13)–C(131) angle is almost 180° thus indicating that the imido group acts as a six-electron donor (sp-hybridisation). In addition to this, the two remaining amido groups at the tantalum centre are almost planar with torsion angles of about 170° supporting the assumption of a strong sp2-double bond character of the Ta(1)–N(14) and Ta(1)–N(15) bonds. Counting the amido groups as four electron donors and the hydrazido ligand as a 2 + 2 electron donor with sp3-hybridized N atoms gives a saturated 18 valence electron count for 5.
Table 2 Selected bond lengths and angles of the crystal structure of 5
| Ta(1)–N(11) |
2.021(7) |
N(11)–Ta(1)–N(12) |
37.6(3) |
| Ta(1)–N(12) |
2.350(7) |
Ta(1)–N(13)–C(131) |
179.6(6) |
| Ta(1)–N(13) |
1.786(7) |
|
|
| Ta(1)–N(14) |
2.013(7) |
Selected torsion angles (°) |
|
| Ta(1)–N(15) |
1.995(7) |
Ta(1)–N(15)–C(151)–C(152) |
170.3/171.2 |
| N(11)–Si(1) |
1.738(7) |
Ta(1)–N(14)–C(141)–C(142) |
172.5/171.3 |
| N(11)–N(12) |
1.443(10) |
Ta(1)–N(11)–N(12)–N(13) |
179.6(4) |
| |
1
|
5
|
8
|
| Formula |
ZrC15H45ClN6Si3 |
TaC13H36N5Si |
C20H40Li2N6Si2 |
|
M
r
|
520.51 |
471.51 |
434.64 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/n |
P21/c |
|
a/Å |
18.019(8) |
9.3189(7) |
9.063(4) |
|
b/Å |
10.197(6) |
33.5381(18) |
19.277(8) |
|
c/Å |
15.690(7) |
13.6538(9) |
8.088(3) |
|
β/° |
94.83(4) |
102.595(6) |
105.113(10) |
|
U/Å3 |
2873(2) |
4164.6(5) |
1364.0(10) |
|
T/K |
105(2) |
105(2) |
203(2) |
|
Z
|
4 |
8 |
2 |
|
µ(Mo-Kα)/mm−1 |
0.612 |
5.336 |
0.146 |
| Reflections collected/unique |
31119/8352 |
32885/9121 |
7796/2408 |
|
R
int
|
0.0551 |
0.0667 |
0.0545 |
|
R
1 (all data) |
0.1009 |
0.0860 |
0.1023 |
|
wR(F2) (all data) |
0.1535 |
0.1213 |
0.1404 |
 |
| | Fig. 3 Molecular structure of 5 in the solid state. | |
Characterisation of 8
The complex [Li(tdmh)(py)]2 was synthesised independently and fully characterised (8), after being identified in reactions of [TaCl3(py)2(N-t-Bu)] with Li(tdmh). Obviously 8 exhibits only low reactivity towards salt metathesis reactions. Even refluxing three equivalents of 8 with [Ta(Cl)3(N-t-Bu)(py)2] for 72 h did not lead to the desired tantalum complex. More equivalents of 8 in these reactions lead to the previously mentioned problems of product isolation and purification. However, compound 8 is interesting for its own merit and we wish to give comment on that here. Depending on the rapidness of addition of the butyllithium to the ligand and the dilution of the compound, the colour of the resulting solution can vary from yellow to red. Nevertheless, no noteworthy impurities can be found in the NMR spectra of 8. It seems though, that an equilibrium of the monomeric and dimeric species of 8 exists in solution, so that two peaks appear for the SiMe3 groups in 1H NMR spectra. These two peaks vary in their intensities, depending on the concentration of the NMR solution. At higher temperatures (60 °C) only one peak can be observed. The pure isolated complex is very air sensitive and decomposes immediately when exposed to air. Single crystals suitable for X-ray structure analysis of 8 were obtained by crystallisation from hexane at −30 °C (Fig. 4). The complex displays a Ci-symmetry with two µ2-η2 coordinated hydrazido ligands. The structure of 8 is very similar to the recently published dimeric complex [Mg(tdmh)2]2.14 Numerous examples of lithium-hydrazido complexes are known and compound 8 matches the typical structural properties.15
 |
| | Fig. 4 Molecular structure of 8 in the solid state. | |
Comparison of the thermal properties of 3–7
Compounds 3–7 are liquids at room temperature that can be quantitatively distilled under reduced pressure. Solid precursors (e.g. the homoleptic trimethylhydrazine complexes of Ti, Zr and Hf) often result in problems to ensure a constant mass transport of the precursor into the gas phase. Therefore, being liquid at ambient temperature simplifies the handling of the precursor. Compounds 3 and 4 display almost the same thermal behaviour in TG/DTA analyses (Fig. 5). Compound 4 seems to be slightly more volatile at lower temperatures, but higher residue is observed. This is due to the higher weight of hafnium, compared to zirconium. Decomposition of the group IV complexes begins at about 202 (3) and 200 °C (4). The two tantalum compounds 5 and 6 are significantly more volatile than the diethyl-substituted complex 7, with the consequence of relatively low remaining masses of 4 and 6%, respectively. Compared to TBTDET the compounds show a higher temperature onset of volatilisation, but they have the advantage of a higher thermal stability. This is revealed by possessing higher decomposition temperature of about 250 °C as against 190 °C for TBTDET. It is to be noted that both, 5 and 6 show monotonic weight loss with almost negligible residue left behind above 250 °C when compared to TBTDET. The DTA spectrum of 7 shows a peak, related to decomposition at a temperature of 280 °C. The high thermal stability of the precursors make them promising candidates for ALD processes, that can be carried out at comparable high temperatures. The higher thermal stability is as expected going along with a lower chemical reactivity of the complexes. Testing these precursors under ALD-conditions would be very interesting, not only in the ALD of metal nitrides, but also in the deposition of thin oxide films, together with water as the oxygen source.
 |
| | Fig. 5 TG spectra of compounds 3, 4 (a) and 5–7 (b). The spectra were recorded at a heating rate of 5 °C min−1 and a nitrogen (purity 6.0) gas flow of 300 sccm. | |
MOCVD Experiments of 7
Compound 7 was selected for MOCVD test experiments because of its liquid nature. The depositions were carried out in a home built horizontal cold wall MOCVD reactor16 at substrate temperatures above 600 °C. The bubbler temperature was kept constant at 140 °C at reduced pressure. In addition to the inert carrier gas nitrogen, ammonia was used as reactive gas in some of the depositions. Best uniformity and lowest roughness of the films were obtained at 700 °C with additional ammonia (Fig. 6). The deposited films were amorphous without significant reflections of TaN in the X-ray diffraction patterns. RBS analysis (Rutherford back scattering) showed that the addition of ammonia significantly reduces the amount of incorporated carbon in the deposited films (Fig. 7). Besides that, no oxygen (below the detection limit) was found in the films. Interestingly, even in the presence of ammonia, Si was detected in the films by RBS measurements. Based on the energy of the collected, back-scattered helium ions, it can be differentiated between the silicon in the film and the silicon from the substrate. The amount of incorporated silicon is relatively high (∼10%) and not expected, especially if one considers the small amount of incorporated carbon. We cannot rigorously rule-out that Si diffuses from the Si(100) substrate through pin-hole defects (not seen by SEM) into the deposited films. But looking at the reported data of TaN thin films grown by MOCVD on Si substrates (with a native SiO2 interface) Si incorporation from the substrate is unlikely. Nevertheless, our preliminary results presented here at least substantiate the potential of the new precursors. More detailed further studies using different substrates and conditions are warranted. These issues are currently under investigation and will be published in a materials properties focussed publication, elsewhere.
![Cross sectional SEM-picture of a TaN(Si) film deposited using [Ta(NEt2)2(N-t-Bu)(tdmh)] (7) at 700 °C with additional ammonia.](/image/article/2006/DT/b512074h/b512074h-f6.gif) |
| | Fig. 6 Cross sectional SEM-picture of a TaN(Si) film deposited using [Ta(NEt2)2(N-t-Bu)(tdmh)] (7) at 700 °C with additional ammonia. | |
 |
| | Fig. 7 RBS spectra of (a) film deposited at 700 °C, with additional 10 sccm of ammonia, (b) film deposited at 600 °C, without ammonia using precursor 7. | |
Conclusion
Based on the considerable interest in the selective formation of thin refractory metal nitride films MN in the metal oxidation state +III, we investigated reactions between hafnium, zirconium and tantalum compounds with hydrazine derivatives. Hydrazine is known for its reductive effect as a reactive gas in MOCVD processes. Therefore, the formation of mixed amido/hydrazido complexes is interesting with respect to mechanistic aspects, since species like this may be relevant intermediates. Experiments with unsymmetric dimethylhydrazine (Hdmh) indicated the high reactivity of the derivative. Transamination reactions were observed in the reactions of Hdmh and the mixed amido/imido compounds of tantalum. These reactions led to the decomposition of the tantalum compounds. Not only alkyl-amido groups, but also alkyl-imido groups were replaced. Thus, further investigations on the mechanism would be helpful to collect more information about the decomposition process of the precursors. In order to isolate stable, hydrazido-ligand containing metal complexes, the ligand Htdmh was introduced. Due to the lack of a second, reactive proton compared to Hdmh, various mixed amido/imido/hydrazido complexes were obtained. The all nitrogen coordinated complexes 3–7 are liquids with adequate volatility, which makes them very good MOCVD precursor candidates. MOCVD experiments carried out with compound 7 as representative example with additional ammonia gave smooth, carbon and oxygen free amorphous TaN layers according to RBS. The origin of about 10% of Si in the films is however still unclear and warrants further investigations.
Experimental
All manipulations of air- and moisture-sensitive compounds were performed employing a conventional vacuum/argon line using standard Schlenk techniques. Sample preparation for further analysis were carried out in argon filled glove boxes. All solvents were purified by an MBraun solvent purification system (SPS) and stored over molecular sieves (4 Å). NMR solvents were degassed and dried over activated molecular sieves. Elemental analysis was performed by the analytical service of our Chemistry Department (CHNSO Vario EL 1998). Proton- and 13C NMR spectra were recorded either on the Bruker Advance DPX-250 (1H: 250.1 MHz, 13C: 62.9 MHz) or the Bruker Advance DPX-200 (1H: 200.1 MHz, 13C: 50.3 MHz). Electronic Ionization Mass spectra were recorded, using a Varian MAT spectrometer. m/z values were referenced to 35Cl, 90Zr, 180Hf and 181Ta. Simultaneous TG/DTA analysis was carried out using a Seiko TG/DTA 6300S11 at ambient pressure (Sample size ∼10 mg). The starting compounds, namely Htdmh, [Zr(NEt2)4], [Hf(NEt2)4], [Ta(N-t-Bu)(py)2(Cl3)], [Ta(NMe2)3(N-t-Bu)], [Ta(NMeEt)3(N-t-Bu)] and [Ta(NEt2)3(N-t-Bu)] were synthesised, following modified literature procedures,17,18 whereas Hdmh and the metal chlorides ZrCl4 (99.5%+) and HfCl4 (98%+) were purchased from Alfa Aesar and used without further purification.
Syntheses
[ZrCl(tdmh)3] 1.
A solution of HN(SiMe3)NMe2 (1.70 g, 12.9 mmol) in hexane (50 cm3) was added dropwise to a cooled (−30 °C) solution of MeLi (8.10 cm3 of a 1.60 molar solution in diethyl ether, 12.9 mmol). The mixture was stirred at room temperature for 4 h and a colorless solution was obtained. This solution was added to a stirred solution of ZrCl4 (1.00 g, 4.29 mmol) in diethyl ether (50 cm3) at −30 °C. The resulting white suspension was allowed to warm slowly to room temperature and stirred for 24 h. The suspension was filtered and the extracts were dried under reduced pressure. A white solid (1.41 g, 63%) was obtained by sublimation at 130 °C/0.001 mbar. Anal. Calc. for C15H45N6Si3ClZr: C 34.60, H 8.73, N 16.15. Found: C 34.92, H 9.07, N 16.44%. Mp 109 °C (uncorrected). δH (25 °C, 200 MHz, CDCl3) 0.18 (27H, s, SiCH3), 2.84 (18H, s, NCH3). δC (25 °C, 50 MHz, CDCl3) 4.0 (SiCH3), 52.3 (NCH3). EI-MS (30 eV): m/z 518 (M+, 2%), 445 (M+
− SiMe3, 2), 387 (M+
− N(SiMe3)NMe2, 4), 132 (HN(SiMe3)NMe2, 42) and 73 (SiMe3, 100).
[HfCl(tdmh)3] 2.
This compound was prepared in a similar method described for 1. A solution of HN(SiMe3)NMe2 (3.63 g, 27.5 mmol) in hexane (50 cm3) was added dropwise to a cooled (−30 °C) solution of MeLi (17.2 cm3 of a 1.60 molar solution in diethyl ether, 27.5 mmol). The mixture was stirred at room temperature for 4 h and a colorless solution was obtained. This solution was added to a stirred solution of HfCl4 (2.00 g, 6.25 mmol) in diethyl ether (50 cm3) at −30 °C. The resulting white suspension was allowed to warm slowly to room temperature and stirred for 24 h. The suspension was filtered and the extracts were dried under reduced pressure. A white solid (2.28 g, 60%) was obtained by sublimation at 150 °C/0.001 mbar. Anal. Calc. for C15H45N6Si3ClHf: C 29.64, H 7.48, N 13.83. Found: C 29.36, H 7.12, N 13.86%. Mp 120 °C (uncorrected). δH (25 °C, 200 MHz, CDCl3) 0.16 (27H, s, SiCH3) and 2.86 (18H, s, NCH3). δC (25 °C, 50 MHz, CDCl3) 3.7 (SiCH3) and 52.6 (NCH3). EI-MS (30 eV): m/z 132 (HN(SiMe3)NMe2, 45%) and 73 (SiMe3, 100).
[Zr(NEt2)3(tdmh)] 3.
HN(SiMe3)NMe2 (0.853 g, 6.45 mmol) was added dropwise to [Zr(NEt2)4] (2.03 g, 5.35 mmol) without any solvent at room temperature. The resulting colorless solution was stirred for 1 h. Volatiles were removed under reduced pressure. A colorless liquid (2.00 g, 85%) was obtained by vacuum distillation. Anal. Calc. for C17H45N5SiZr: C 46.52, H 10.33, N 15.96. Found: C 43.43, H 10.42, N 16.34. (Large deviation of the carbon value due to problems with the carbon column) δH (25 °C, 200 MHz, C6D6) 0.25 (9H, s, SiCH3), 1.15 (18H, t, NCH2CH3), 2.52 (6H, s, NCH3) and 3.40 (12H, q, NCH2CH3). δC (25 °C, 50 MHz, C6D6) 3.4 (SiCH3), 16.2 (NCH2CH3), 44.7 (NCH2CH3) and 52.2 (NCH3). EI-MS (30 eV): m/z 437 (M+, 2%), 306 (Zr(NEt2)3, 100), 234 (Zr(NEt2)2, 70), 162 (Zr(NEt2), 90) and 73 (SiMe3, 40).
[Hf(NEt2)3(tdmh)] 4.
This compound was prepared by the method described for 3. HN(SiMe3)NMe2 (0.860 g, 6.50 mmol) was added dropwise to [Hf(NEt2)4] (2.51 g, 5.39 mmol) without any solvent at room temperature. The resulting colorless solution was stirred for 1 h. Volatiles were removed under reduced pressure. A colorless liquid (1.97 g, 69%) was obtained by vacuum distillation. Anal. Calc. for C17H45N5SiHf: C 38.81, H 8.62, N 13.31. Found: C 38.28, H 9.76, N 13.83. δH (25 °C, 200 MHz, C6D6) 0.23 (9H, s, SiCH3), 1.16 (18H, t, NCH2CH3), 2.52 (6H, s, NCH3), 3.42 (12H, q, NCH2CH3). δC (25 °C, 50 MHz, C6D6) 3.5 (SiCH3), 16.2 (NCH2CH3), 44.4 (NCH2CH3), 52.0 (NCH3); EI-MS (30 eV): m/z 527 (M+, 1%), 454 (M+
− SiMe3, 2), 396 (Hf(NEt2)3, 1) and 73 (SiMe3, 40).
[Ta(NMe2)2(tdmh)(NtBu)] 5.
Compound 5 was synthesised by adding 5.78 g (43.7 mmol) Htdmh to 14.0 g (36.4 mmol) [Ta(NMe2)3(N-t-Bu)], dissolved in 20 cm3 hexane. The slightly yellow solution was stirred for 48 h at room temperature. After all volatiles were removed in vacuo, the colourless, viscous liquid (5) was obtained by distillation at 135 °C, 0.01 mbar. Crystals, suitable for single-crystal X-ray diffraction were obtained from a high concentrated solution of 5 in hexane at −20 °C. Yield 13.1 g, 77% (27.8 mmol) (based on [Ta(NMe2)3(N-t-Bu)]), Anal. Calc. for C13H36N5SiTa: C 33.12, H 7.70, N 14.85. Found: C 33.15, H 7.49, N 15.20%. δH (25 °C, 250 MHz, C6D6) 0.31 (9H, s, NSi(CH3)3), 1.52 (9H, s, NC(CH3)3), 2.41 (6H, s, N–N(CH3)2), 3.40 (12H, s, Ta–N(CH3)2). δC (25 °C, 62.5 MHz, C6D6) 3.8 (NSi(CH3)3), 34.7 (NC(CH3)3), 49.3 (N–N(CH3)2), 51.3 (Ta–N(CH3)2), 64.3 (NC(CH3)3). EI-MS (70 eV): m/z 471 (M+, 3%), 456 (M+
− CH3, 8), 73 (SiMe3, 12), 58 (H-t-Bu, 77), 44 (NMe2, 100).
[Ta(NMeEt)2(tdmh)(NtBu)] 6.
The preparation of 6 follows exactly the synthesis route of 5. Instead of [Ta(NMe2)3(N-t-Bu)] as the metal containing starting compound, [Ta(NMeEt)3(N-t-Bu)] (14.8 g, 34.0 mmol) was used in the synthesis. An amount of 14.8 g (40.8 mmol) Htdmh was added. The colourless, viscous liquid (6) was obtained by distillation at 140 °C, 0.01 mbar. Yield 14.1 g, 83% (28.2 mmol) (based on [Ta(NMeEt)3(N-t-Bu)]), Anal. Calc. for C15H40N5SiTa: C 36.02, H 8.07, N 14.02. Found: C 36.00, H 7.65, N 14.33%. δH (25 °C, 250 MHz, C6D6) 0.32 (9H, s, NSi(CH3)3), 1.23 (12H, t, N(CH3)CH2CH3), 1.49 (9H, s, NC(CH3)3), 2.42 (6H, s, N–N(CH3)2), 3.15 (6H, s, Ta–N(CH3)CH2CH3), 3.68–3.86 (4H, m, N(CH3)CH2CH3). δC (25 °C, 62.5 MHz, C6D6) 3.8 (NSi(CH3)3), 16.9 (N(CH3)CH2CH3), 34.7 (NC(CH3)3), 42.8 (N(CH3)CH2CH3), 51.3 (N–N(CH3)2), 57.1 (N(CH3)CH2CH3), 64.3 (NC(CH3)3). EI-MS (70 eV): m/z 499 (M+, 2%), 484 (M − CH3, 5), 73 (SiMe3, 12), 58 (H-t-Bu, 76), 44 (NMe2, 100).
[Ta(NEt2)2(tdmh)(NtBu)] 7.
The preparation of 7 on the whole follows the synthesis route employed for 5 and 6. Instead of [Ta(NMe2)3(N-t-Bu)] as the metal containing starting compound, [Ta(NEt2)3(N-t-Bu)] (9.23 g, 19.7 mmol) was used in the synthesis. In this reaction, a high excess of Htdmh (10.41 g, 78.7 mmol) was added. The mixture was refluxed for 5 days. Unreacted ligand and the solvent was removed in vacuo and the colourless, viscous liquid (5) was obtained by distillation at 143 °C, 0.01 mbar. Yield 9.2 g, 88% (17.3 mmol) (based on [Ta(NEt2)3(N-t-Bu)]), Anal. Calc. for C17H44N5SiTa: C 38.70, H 8.41, N 13.27. Found: C 38.50, H 8.27, N 13.65%. δH (25 °C, 250 MHz, C6D6) 0.34 (9H, s, NSi(CH3)3), 1.12 (12H, t, NCH2CH3), 1.49 (9H, s, NC(CH3)3), 2.43. δC (25 °C, 62.5 MHz, C6D6) 3.8 (NSi(CH3)3), 17.4 (NCH2CH3), 34.7 (NC(CH3)3), 49.2 (NCH2CH3), 51.3 (N–N(CH3)2), 64.3 (NC(CH3)3). EI-MS (70 eV): m/z 527 (M+, 40%), 512 (M − CH3, 100), 441 (M − (N-t-Bu) − CH3, 38), 73 (SiMe3, 23), 58 (H-t-Bu, 60), 44 (NMe2, 100).
[Li(tdmh)py]28.
Compound 8 was synthesised by adding n-butyllithium (15.4 mmol, 9.68 cm3 of a 1.60 molar solution in hexane) to a cooled solution (−78 °C) of Htdmh (2.04 g, 15.4 mmol) and pyridine (5.5 cm3, 67.5 mmol) in 5 cm3 hexane. The solution turns red after warming to room temperature. The solution is stirred for another 24 h and then cooled to −30 °C. The compound crystallises in the form of small, light brown crystals. Mp 93 °C (uncorrected), yield 2.2 g, 64% (based on Htdmh), M = 434.6 g mol−1, Anal. Calc. for C20H40N6Si2Li2: C 55.27, H 9.28, N 19.34. Found: C 55.25, H 9.42, N 20.09%. δH (25 °C, 250 MHz, C6D6) 0.36, 0.43 (18H, s, NSi(CH3)3), 2.67 (12H, s, N–N(CH3)2), 6.63 (4H, d, o-C5H5N), 6.91 (1H, t, p-C5H5N), 8.64 (4H, t, m-C5H5N). δC (25 °C, 62.5 MHz, C6D6) 5.7, 6.0 (NSi(CH3)3), 53.8 (N–N(CH3)2), 133.0 (o-C5H5N), 136.6 (p-C5H5N), 149.9 (m-C5H5N). EI-MS (24 eV): m/z 284 (M+
− pyridine − SiMe3, 8%), 132 (Htdmh, 26), 117 (Htdmh − Me, 17), 79 (pyridine, 100), 73 (SiMe3, 37), 52 (HNMe2Li, 28%).
X-Ray crystallography
Single-crystal diffraction experiments were carried out at 105 K (1 and 5) and 203 K (8), respectively on an Oxford Xcalibur 2 CCD/PD Diffractometer (1 and 5) or a Bruker-AXS-SMART (CCD 1000) Diffractometer (8) with monochromated Mo-Kα radiation (0.71073 Å). The structures were solved by direct methods and refined anisotropically with SHELXL-97 program suite. Crystal data of 1, 5 and 8 are listed in Table 3.
CCDC reference numbers 282095–282097.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512074h
Acknowledgements
The authors would like to thank the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie and H. C. Starck for financial support.
References
- L. Peters, Semicond. Int., 2003, 263, 50–52 Search PubMed; H. Y. Yu, H. F. Lim, J. H. Chen, M. F. Li, C. Zhu, C. H. Tung, A. Y. Du, W. D. Wang, D. Z. Chi and D.-L. Kwong, IEEE Electron Device Lett., 2003, 24, 230 CrossRef CAS; H. Y. Yu, J. F. Kang, C. Ren, J. D. Chen, Y. T. Hou, S. Shen, M. F. Li, D. S. H. Chan, K. L. Bera, C. H. Tung and D.-L. Kwong, IEEE Electron Device Lett., 2004, 25, 70 CrossRef CAS.
- A. Ishikawa, T. Takata, J. N. Kondo, M. Hara and K. Domen, J. Phys. Chem. B, 2004, 108, 11049–11053 CrossRef CAS.
-
M. Ritala, in High-k Gate Dielectrics, ed. M. Houssa, Institute of Physics Publishing, Bristol, 2004, pp. 17–64 Search PubMed.
- A. Baunemann, D. Rische, A. Milanov, Y. Kim, M. Winter, C. Gemel and R. A. Fischer, Dalton Trans., 2005, 3051–3056 RSC and references therein; C. H. Winter, Aldrichim. Acta, 2000, 33, 3–12 Search PubMed.
- J. E. Bleau, C. J. Carmalt, S. A. O'Neill, I. P. Parkin, A. J. P. White and D. J. Williams, Polyhedron, 2005, 24, 463–468 CrossRef CAS.
- W. Wang, T. Nabatame and Y. Shimogaki, Jpn. J. Appl. Phys., 2004, 43(Part 2), L1445–L1448 CrossRef CAS; R. Fix, R. G. Gordon and D. M. Hoffman, Chem. Mater., 1991, 3, 1138–1148 CrossRef CAS; D. M. Hoffman, Polyhedron, 1994, 13, 1169–1179 CrossRef CAS; R. Fix, R. G. Gordon and D. M. Hoffman, Chem. Mater., 1993, 5, 614–619 CrossRef CAS.
- B. Luo, C. J. Cramer and W. L. Gladfelter, Inorg. Chem., 2003, 42, 3431–3437 CrossRef CAS.
- J.-S. M. Lehn and D. M. Hoffman, Inorg. Chim. Acta, 2003, 345, 327–332 CrossRef CAS.
- C. H. Winter, K. C. Jayaratne and J. W. Proscia, Mater. Res. Soc. Symp. Proc., 1994, 327, 103–108 CAS;
C. H. Winter, T. S. Lewkebandara and K. C. Jayaratne, US Pat., 5,591,483, 1994 Search PubMed.
- M. Juppo, M. Ritala and M. Leskela, J. Electrochem. Soc., 2000, 147, 3377–3381 CAS; K. T. Jacob, R. Verma and R. M. Mallya, J. Mater. Sci., 2002, 37, 4465–4472 CrossRef CAS; D. K. Gaskill, N. Bottka and M. C. Lin, J. Cryst. Growth, 1986, 77, 418–423 CrossRef CAS; D. A. Wierda and C. Armado-Wierda, Proc. Electrochem. Soc., 2000, 2000–13(CVD XV), 497–504 CAS.
- Y.-S. Suh, G. P. Heuss, V. Misra, D.-G. Park and K.-Y. Lim, J. Electrochem. Soc., 2003, 150, F79–F82 CrossRef CAS; K. Shepherd and J. Kelber, Appl. Surf. Sci., 1999, 151, 287–298 CrossRef CAS; H.-L. Park, K.-M. Byun and W. J. Lee, Jpn. J. Appl. Phys., 2002, 41, 6153–6164 CrossRef CAS.
- Y. Kim, A. Baunemann, H. Parala, A. Devi and R. A. Fischer, Chem. Vap. Deposition, 2005, 11, 294–297 CrossRef CAS.
- A. S. Batsanov, A. V. Churakov, J. A. K. Howard, A. K. Hughes, A. L. Johnson, A. J. Kingsley, I. S. Neretin and K. Wade, J. Chem. Soc., Dalton Trans., 1999, 3867–3875 RSC.
- H. Sachdev, Eur. J. Inorg. Chem., 2002, 2681–2685 CrossRef CAS.
- N. Metzler, H. Noth and H. Sachdev, Angew. Chem., Int. Ed. Engl., 1994, 33, 1746 CrossRef; K. Bode, U. Klingebiel, M. Noltemeyer and H. Witte-Abel, Z. Anorg. Allg. Chem., 1995, 621, 500 CAS; B. Germund, H. Noth, H. Sachdev and M. Schmidt, Chem. Ber., 1996, 129, 1335 CrossRef.
- A. Devi, W. Rogge, A. Wohlfart, F. Hipler, H.-W. Becker and R. A. Fischer, Chem. Vap. Deposition, 2000, 6, 245–252 CrossRef CAS.
- H.-T. Chiu, S.-H. Chuang, C.-E. Tsai, G.-H. Lee and S.-M. Peng, Polyhedron, 1998, 17, 2187–2190 CrossRef CAS; W. A. Nugent, Inorg. Chem., 1983, 22, 965–969 CrossRef CAS; D. C. Bradley and M. H. Gitlitz, J. Chem. Soc. A, 1969, 980–984 RSC.
- U. Wannagat and F. Hoefler, Monatsh. Chem., 1966, 97, 976–983 CAS.
|
| This journal is © The Royal Society of Chemistry 2006 |
Click here to see how this site uses Cookies. View our privacy policy here.