Supramolecular architectures assembled by the interaction of purine nucleobases with metal-oxalato frameworks. Non-covalent stabilization of the 7H-adenine tautomer in the solid-state†
Juan P.
García-Terán
,
Oscar
Castillo
*,
Antonio
Luque
*,
Urko
García-Couceiro
,
Garikoitz
Beobide
and
Pascual
Román
Departamento de Química Inorgánica, Facultad de Ciencia y Tecnología, Universidad del País Vasco, Apartado 644, E-48080 Bilbao, Spain. E-mail: oscar.castillo@ehu.es; antonio.luque@ehu.es; Fax: +34 94 601 3500; Tel: +34 94 601 5991
First published on 5th December 2005
Abstract
The synthesis, crystal structure and variable-temperature magnetic measurements of the compounds [Mn(µ-ox)(H2O)(7H-pur-κN9)]n (1), {[Mn(µ-ox)(H2O)2]·(7H-ade)·(H2O)}n (2) and {[Cu(µ-ox)(H2O)(7H-ade-κN9)][Cu(µ-ox)(µ-H2O)(7H-ade-κN9)]·∼10/3H2O}n (3), (where ox: oxalato dianion, pur: purine, and ade: adenine) are reported. Compounds 1 and 2 contain one-dimensional chains in which manganese(II) atoms are bridged by bis-bidentate oxalato ligands. The distorted octahedral geometry around each metal centre is completed in compound 1 by one water molecule and the imidazole N9 donor site of the purine ligand, which is a rare example of direct binding between the Mn(II) ion and the N donor site of an isolated nucleobase. Unlike 1, the adenine moiety in compound 2 is not bonded to manganese atoms and the metal coordination polyhedron is filled by two water molecules in a cis-arrangement. Its crystal building is constructed from π-stacked layers of Watson–Crick hydrogen-bonded adenine⋯(H2O)2⋯adenine aggregates and zig–zag Mn(II)-oxalato chains held together by means of a strong network of hydrogen bonding interactions. The nucleobase exists in the lattice as the 7H-adenine tautomer which represents an unprecedented solid-state characterization of this minor tautomer as free molecule (without metal coordination) stabilized through non-covalent interactions. Compound 3 consists of two slightly different [Cu(ox)(H2O)(7H-ade-κN9)] units in which the nucleobase coordinates through the imidazole N9 atom. The planar complex entities are parallel stacked and joined by means of long Cu–O bonds involving oxygen atoms from the oxalato and the aqua ligands, giving one-dimensional chains with a [4 + 1] square-planar pyramidal and a [4 + 2] octahedral coordination around the metal centre, respectively. Self-assembled process of compound 3 is further driven by an in-plane network of hydrogen bonding interactions to generate a porous 3D structure containing parallel channels filled by guest water molecules. Variable-temperature magnetic susceptibility measurements of all the complexes show the occurrence of antiferromagnetic interactions between the paramagnetic centres. DFT calculations have been performed to check the influence of packing in the stability of the 7H-amino tautomer of 2 and in the complex geometry of 3.
Introduction
After the discovery of the famous DNA double-stranded helix by Watson and Crick1 many research efforts have been dedicated to the rational design and elaboration of biomimetic systems based on the interaction of nucleobases and its derivatives with a wide range of metal ions.2–3 Studies into metal–nucleobase bindings are of great interest since metal ions play a crucial role in the structure and function of nucleic acids and genetic information transfer.4 The binding preferences of a specific metal ion towards nucleic acids or their constituents depends essentially on the metal properties (main group or transition-metal, charge, d-electron configuration, and hard- or softness), the basicity of the N/O donor site of the nucleobases and, sometimes, the auxiliary ligands which complete the metal coordination sphere.5 On the other hand, the non covalent forces which govern the supramolecular assemblies of these biomimetic systems (such as hydrogen bonds and arene–arene π-stacking) are operative in the control of molecular recognition processes involving a great diversity of macromolecular biological systems.6 So that, the synthesis of such systems, coupled with a fairly detailed understanding of the chemical and physical properties of both covalent and non covalent interactions7–11 offers a powerful tool for the development of advanced functional materials12 with potential applications for medical and molecular sciences.13Among the nucleobases, adenine (6-aminopurine) shows the widest range of binding possibilities2,14 because it exhibits at least five donor sites (N9, N7, N3, N1 and N6), and a great variety of complexes with different metal-ion binding patterns have been reported [see ESI†].15 The purine itself, without the exo-cyclic N6 amino group, also possesses a great coordination versatility but a scarce number of complexes has been structurally characterized so far, probably because it is not usually present in biopolymers.16–18 Most of the transition-metal complexes of both purine bases are monomers or discrete oligomeric species and the examples of n-dimensional (nD, n = 1–3) coordination networks based solely on covalent bonds are quite limited16,17,19,20 despite the high scientific interest that such complexes, resembling DNA dimensionality, might show. In this field, we have recently reported the structural and magnetic characterization of a new family of oxalato-bridged compounds with formulae [M(µ-ox)(H2O)(pur)]n and {[M(µ-ox)(H2O)(ade)]n·2(ade)·(H2O)}n (where M(II) is Cu, Co, and Zn) based on 1D metal-oxalato framework in which the nucleobase behaves as monodentate N9/N3-bound ligand.21
Following our current research on metal-nucleobase systems, we report herein the synthesis of the polymeric compounds [Mn(µ-ox)(H2O)(7H-pur-κN9)]n (1), {[Mn(µ-ox)(H2O)2]·(7H-ade)·(H2O)}n (2), and {[Cu(µ-ox)(H2O)(7H-ade-κN9)][Cu(µ-ox)(µ-H2O)(7H-ade-κN9)]·∼10/3H2O}n (3), and their characterization by X-ray crystallography, FT-IR spectroscopy and variable-temperature magnetic susceptibility measurements. Although there are reported examples of Mn(II) binding to N donor sites of nucleobases in biopolymeric systems, coordinative Mn–N linkages involving isolated nucleobases, as seen in 1, are scarce.22,23 In fact, in complex 2 the supramolecular assembly does not evolve towards a molecule containing direct binding of the metal to the nucleobase. Of particular note, is the fact that the neutral adenine nucleobase occurs in its tautomeric 7H-amino form. Scheme 1 shows the four ground state tautomers of the amino form of the adenine molecule. As far as we are aware, compound 2 represents the first crystallographic characterization (in solid form) of the minor 7H-adenine tautomer as uncomplexed molecule since it has been found usually in compounds where the proton transfer is brought about by a synergy of metal coordination to the N9 donor site and an efficient stabilization of the 7H-adenine ligand by hydrogen bonding interactions,24,25 as occurs in compound 3. Compound 3 exhibits two different planar complex entities in which the adenine ligand is bound to the Cu(II) centres through the N9 donor site of the imidazole ring and the proton is shifted to the N7 position. Square-planar transition-metal complexes containing purine nucleobases (or ligands with aromatic rings) are of increasing importance in biotechnology and medicine because several of them can bind to duplex DNA through intercalation into the DNA base stack.2,3
 | ||
| Scheme 1 | ||
Experimental
Materials
All chemicals were of reagent grade and were used as commercially obtained. Standard literature procedures were used to prepare the starting materials K2[Cu(ox)2]·2H2O and [Mn(µ-ox)(H2O)2]n.26Preparations
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C); 1483s (δ(C2–H + C8–N9) + ν(C8–H)); 1423m, 1405s δ(N1–C6–H6); 1368m ν(C5–N7–C8); 1313s (ν(N9–C8 + N3–C2) + δ(C–H) + νs(O–C–O)); 1265s (δ(C8–H) + ν(N7–C8)); 978w, 964m ν(N1–C6); 801s δ(O–C–O); 680m, 651m, 608m (ring deformation); 577m, 513m, 500w and 462m ν(M–O + M–N).
C) + δ(NH2)); 1564sh ν((C4–C5) + (N3–C4–C5)); 1507w, 1489m (δ(C2–H + C8–N9) + ν(C8–H)); 1448m δ(N1–C6–H6); 1398m, 1359m ν(C5–N7–C8); 1320s (ν(N9–C8 + N3–C2) + δ(C–H) + νs(O–C–O)); 1252m, 1234w, 1164m (δ(C8–H) + ν(N7–C8)); 1123m, 1023m τ(NH2); 945m, 911m (ν(N1–C6) + τ(NH2)); 798s, 789s δ(O–C–O); 716m (ring deformation); 573m and 534s ν(M–O + M–N).
C) + δ(NH2)); 1616s (ν(CC) + δ(NH2)); 1570m ν((C4–C5) + (N3–C4–C5)); 1515m, 1482m (δ(C2–H + C8–N9) + ν(C8–H)); 1427s, 1397m δ(N1–C6–H6); 1360w ν(C5–N7–C8); 1333m (ν(N9–C8 + N3–C2) + δ(C–H) + νs(O–C–O)); 1265m, 1214m (δ(C8–H) + ν(N7–C8)); 1030w τ(NH2); 977w, 915w (ν(N1–C6) + τ(NH2)); 793m δ(O–C–O); 720w, 691w (ring deformation); 609w, 565sh, 538m and 481w ν(M–O + M–N).
C); 1483s (δ(C2–H + C8–N9) + ν(C8–H)); 1423m, 1405s δ(N1–C6–H6); 1368m ν(C5–N7–C8); 1313s (ν(N9–C8 + N3–C2) + δ(C–H) + νs(O–C–O)); 1265s (δ(C8–H) + ν(N7–C8)); 978w, 964m ν(N1–C6); 801s δ(O–C–O); 680m, 651m, 608m (ring deformation); 577m, 513m, 500w and 462m ν(M–O + M–N).
C) + δ(NH2)); 1564sh ν((C4–C5) + (N3–C4–C5)); 1507w, 1489m (δ(C2–H + C8–N9) + ν(C8–H)); 1448m δ(N1–C6–H6); 1398m, 1359m ν(C5–N7–C8); 1320s (ν(N9–C8 + N3–C2) + δ(C–H) + νs(O–C–O)); 1252m, 1234w, 1164m (δ(C8–H) + ν(N7–C8)); 1123m, 1023m τ(NH2); 945m, 911m (ν(N1–C6) + τ(NH2)); 798s, 789s δ(O–C–O); 716m (ring deformation); 573m and 534s ν(M–O + M–N).
C) + δ(NH2)); 1616s (ν(CC) + δ(NH2)); 1570m ν((C4–C5) + (N3–C4–C5)); 1515m, 1482m (δ(C2–H + C8–N9) + ν(C8–H)); 1427s, 1397m δ(N1–C6–H6); 1360w ν(C5–N7–C8); 1333m (ν(N9–C8 + N3–C2) + δ(C–H) + νs(O–C–O)); 1265m, 1214m (δ(C8–H) + ν(N7–C8)); 1030w τ(NH2); 977w, 915w (ν(N1–C6) + τ(NH2)); 793m δ(O–C–O); 720w, 691w (ring deformation); 609w, 565sh, 538m and 481w ν(M–O + M–N).
Physical characterisation
Elemental analyses (C, H, N) were performed on a Perkin-Elmer 2400 microanalytical analyser. Metal content was determined by absorption spectrometry. The IR spectra (KBr pellets) were recorded on a FTIR Mattson 1000 spectrometer in the 4000–400 cm−1 spectral region. Magnetic measurements were performed on polycrystalline samples of the complexes, taken from the same uniform batches used for the structural determinations, with a Quantum Design SQUID susceptometer covering the temperature range 2.0–300 K at a magnetic field of 1000 G. The susceptibility data were corrected for the diamagnetism estimated from Pascal's Tables,27 the temperature-independent paramagnetism and the magnetization of the sample holder.X-Ray analysis and structure refinement
Diffraction data were collected at 293(2) K on an Oxford Xcalibur diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Indexation and unit cell refinement were based on all observed reflections from 10 frames collected with an oscillation range of 1° frame−1 and an exposure time of 3 min frame−1. The data reduction was performed with the CrysAlis RED program.28 Structures were solved by direct methods using the SIR92 program29 and refined by full-matrix least-squares on F2 including all reflections (SHELXL97).30 All non-hydrogen atoms were refined anisotropically, except those belonging to the crystallization water molecules of compound 3. These solvent molecules show a pronounced disorder, and fractional oxygens were fitted to the most dominant electron density peaks. Occupancy factors were tentatively refined, and in the final cycles kept fixed at values close to those obtained in the refinement. H atoms of 1 and 2 could be located in the difference Fourier synthesis map and their positions were refined. Hydrogen atoms of ligands in 3 were included in the final cycles of the refinement using a riding model. All calculations were performed using the WINGX crystallographic software package.31 The final geometrical calculations and the graphical manipulations were carried out with the PARST9532 and PLATON33 programs.Computational details
All the quantum calculations of geometry optimization of the compounds and the total dipolar moment of the 9H- and 7H-adenine tautomers have been carried out using the density functional theory with Becke's three-parameter exchange functional34 along with the Lee–Yang–Parr non-local correlation functional (B3LYP).35 The standard 6-31G(d) basis set was used as implemented in the Gaussian03 program.36 The initial geometry of the models was built from the experimental crystal structures.Results and discussion
Description of structures
| 1 | 2 | 3 | |
|---|---|---|---|
| a R1 = ∑‖Fo| − |Fc‖/∑|Fo|; wR2 = [∑[w(Fo2 − Fc2)2]/[∑(Fo2)2]]1/2 where w = 1/[σ2(Fo2) + (AP)2] with A = 0.0196 (1), 0.0172 (2), and 0.0308 (3). | |||
| Empirical formula | C7H6MnN4O5 | C7H11MnN5O7 | C14H20.66Cu2N10O13.33 |
| M r | 281.10 | 332.15 | 669.24 |
| Crystal system | Monoclinic | Triclinic | Triclinic |
| Space group | P21/a |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å | 6.913(1) | 7.340(1) | 3.587(1) |
| b/Å | 16.283(3) | 9.513(2) | 17.965(2) |
| c/Å | 8.594(2) | 9.741(2) | 18.010(2) |
| α/° | 90 | 75.47(2) | 97.70(1) |
| β/° | 105.09(2) | 80.60(2) | 92.42(1) |
| γ/° | 90 | 75.17(2) | 91.87(1) |
| V/Å3 | 934.0(3) | 632.9(2) | 1148.2(4) |
| Z | 4 | 2 | 2 |
| D c/Mg m−3 | 1.999 | 1.743 | 1.936 |
| Color | Colorless | Colorless | Blue |
| Crystal habit | Polyhedric | Polyhedric | Prismatic |
| Crystal size/mm3 | 0.13 × 0.12 × 0.07 | 0.09 × 0.06 × 0.02 | 0.07 × 0.04 × 0.01 |
| µ/mm−1 | 1.433 | 1.085 | 1.944 |
| 2θmax/° | 56.18 | 56.12 | 56.12 |
| Completeness to θmax (%) | 99.8 | 99.0 | 99.3 |
| Reflections collected | 7058 | 5463 | 9791 |
| Independent reflections | 2272 | 3036 | 5540 |
| R int | 0.0345 | 0.0337 | 0.0548 |
| Variable parameters | 172 | 214 | 350 |
| R1/wR2a | 0.0317/0.0555 | 0.0362/0.0572 | 0.0573/0.1302 |
| Goodness-of-fit (F2) | 1.006 | 1.035 | 1.119 |
| Δρmax/min/e Å−3 | 0.421, −0.268 | 0.481, −0.253 | 0.725, −1.350 |
| 1 | 2 | |
|---|---|---|
| L = N(9) for 1 and O(6w) for 2. | ||
| Mn–O(1) | 2.173(2) | 2.209(2) |
| Mn–O(2) | 2.156(2) | 2.183(2) |
| Mn–O(3) | 2.217(2) | 2.218(2) |
| Mn–O(4) | 2.175(2) | 2.182(2) |
| Mn–O(5w) | 2.174(2) | 2.110(2) |
| Mn–L | 2.253(2) | 2.126(2) |
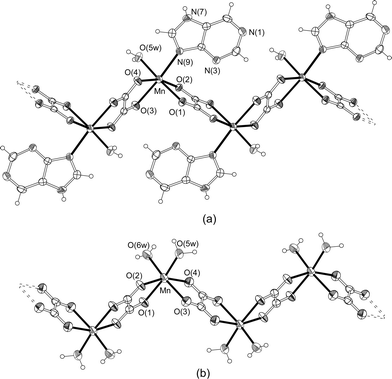 | ||
| Fig. 1 Perspective drawings of the polymeric chains of 1 (a) and 2 (b). Thermal ellipsoids are drawn at the 50% probability level. | ||
The distorted octahedral geometry around the Mn(II) atom (placed on a general position) is completed in compound 1 by one water molecule and the imidazole N9 atom of the purine, giving rise to a cis-MnO4OwN donor set. Nevertheless, in the crystal building of 2, the adenine nucleobase remains free and the metal coordination polyhedron is filled by two water molecules, resulting in a cis-MnO4(Ow)2 chromophore. It is important to note that the cis disposition of the terminal ligands in both polymeric chains implies the rupture, during the synthesis process, of the starting [Mn(µ-ox)(H2O)2]n compound in which the two water molecules are coordinated in trans disposition with respect to the linear manganese-oxalato framework.37
One of the interesting structural aspects of 1 is the direct metal binding to the N atom of the purine base. Manganese(II)–nucleobase bonds involving N donor sites are common in biological systems10 but, as far as we are aware, they are extremely rare in structurally characterized coordination compounds containing isolated nucleobases. Mn(II) is used as a substitute for Mg(II) in supporting nucleic acid folding and function in vitro40,41 and it has often been employed to investigate potential metal binding sites in nucleobases. Nevertheless, the coordination details may differ between the two ions42 owing to Mg(II) and high-spin Mn(II) are similar but not identical in properties (such as ionic radius, enthalpy of hydration, and coordination preferences).43 For example, Mn(II) replaced Mg(II) at all sites that included innersphere coordination to the complex folded structure of the group I P4–P6 RNA, but the indirect interactions through aqua ligands of the hydrated Mg(II) ions are only partially replaced by direct coordination between the Mn and phosphate or nucleic acid base positions.41 A 10-nt RNA duplex containing sheared adenine : guanine basepairs44 and the hammer-head ribozyme45 were shown to have a similar Mn(II) binding pattern involving the phosphate oxygen of the adenosine residue and the N7 position of the preceding guanine residue.
Nevertheless, a search in the Cambridge Structural Database (CSD, release May 2005)15 revealed that the nucleotide complexes [Mn(H2O)5(5-GMP)]·2H2O (5-GMP: guanosine-5′-monophosphate)22 and [Mn(H2O)5(5-IMP)]·2H2O (5-IMP: inosine-5′-monophosphate)23 are the only structurally characterized examples where an isolated nucleobase is coordinated to the Mn(II) ion through N donor sites. Mn(II) coordinates the guanine N7 site and five water molecules with through-water interactions to the exocyclic carbonyl O6 atom and the phosphate group of the nucleotide. The Mn–N bond distance in 1 [2.253(2) Å] is somewhat shorter than those found in the two nucleotides (ranging from 2.318(4) to 2.366(4) Å), but it agrees very well with the values reported for the Mn(II) ion bonded to biological systems (ca. 2.2 Å). Interestingly, the Mn–ATP and Mn–CMP complexes only show chelation of the metal with oxygen atoms of the phosphate group and/or the cytosine residue.46 Furthermore, to date, there was only one case of manganese(II) complex containing non-substituted adenine, the monomer {[Mn(quin2-c)2(H2O)2]·2(9H-ade)} (where quin2-c = quinoline-2-carboxylato), and the nucleobase, with only N donor sites, is out of the coordination sphere of the metal ion.47 Based upon these data, the existence of solely uncomplexed adenine molecules in the crystal building of compound 2 is not surprising.
Obviously, the distinctive coordination abilities of the purine and the adenine with respect to the Mn(II) ion results in a dissimilar organization of the molecular building groups, and as a consequence, a completely different self-assembled supramolecular architecture for compounds 1 and 2. The crystal packing of 1 is similar to that described for the analogous Co(II) and Zn(II) complexes.21 Polymeric chains running along the [101] direction are joined by means of short O5w–H52w⋯O3 (ox2) hydrogen bonds giving rise to sheets which spread out along the crystallographic ac-plane (Fig. 2). The purine ligands are projected forward to the outside of the layers along the b-direction and cross-link adjacent sheets by means of two hydrogen bonds: O5w–H51w⋯N1 (as acceptor) and N7–H7⋯O2 (as donor). The overall 3D supramolecular structure is also stabilized by significant offset face-to-face π–π stacking interactions between the aromatic rings of purine bases belonging to adjacent layers with a centroid–centroid vector of 3.5 Å and an interplanar distance of around 3.3 Å.
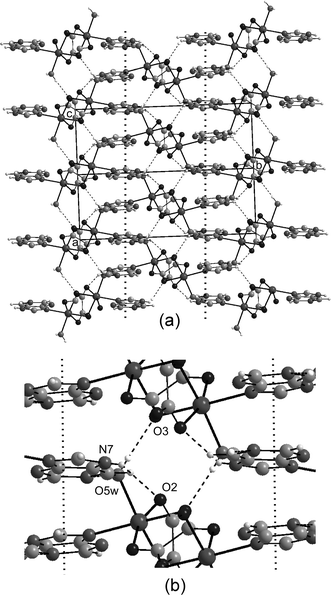 | ||
| Fig. 2 (a) Hydrogen bonding (dashed) and π–π-stacking (dotted line) interactions in the crystal packing of compound 1. (b) Closed view of the interchain interactions. | ||
With respect to compound 2, the nucleobase forms centrosymmetric adenine⋯(H2O)2⋯adenine aggregates (Fig. 3) in which the Watson–Crick edges (N1 and N6H) of each adenine interact with two symmetry-related lattice water molecules by means of strong hydrogen bonds (Table 3). The two adenine moieties and the water molecules are essentially coplanar and the N6-H61 group acts as hydrogen-bond donor to the O7w water molecule, which in turn is bonded to the N1 acceptor from the symmetry centre related moiety.
| D–H⋯Ab | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|
| 1 | |||
| O(5w)–H(51w)⋯N(1)i | 1.98(2) | 2.775(3) | 179(3) |
| O(5w)–H(52w)⋯O(3)ii | 2.03(2) | 2.792(2) | 169(3) |
| N(7)–H(7)⋯O(2)iii | 2.20(2) | 2.834(3) | 147(3) |
| 2 | |||
|---|---|---|---|
| a Symmetry codes: (i) 1/2 + x, 1/2 − y, 1 + z; (ii) 1 − x, −y, 2 − z; (iii) 1/2 + x, 1/2 − y, z; (iv) x, −1 + y, 1 + z; (v) 1 − x, −y, 1 − z; (vi) 1 − x, 1 − y, −z; (vii) −x, 1 − y, 1 − z; (viii) 1 + x, y, z; (ix) x, y, 1 + z. b D: donor, A: acceptor. | |||
| O(5w)–H(51w)⋯N(9)iv | 1.75(3) | 2.719(3) | 169(2) |
| O(5w)–H(52w)⋯O(7w) | 1.98(3) | 2.826(3) | 177(3) |
| O(6w)–H(61w)⋯O(4)v | 2.12(3) | 2.831(2) | 168(3) |
| O(6w)–H(62w)⋯N(3)vi | 1.85(3) | 2.771(3) | 178(2) |
| N(6)–H(61)⋯O(7w)v | 2.22(3) | 3.003(3) | 161(2) |
| N(6)–H(62)⋯O(1) | 2.16(2) | 3.005(3) | 166(2) |
| N(7)–H(7)⋯O(2)vii | 1.94(3) | 2.773(3) | 161(2) |
| O(7w)–H(71w)⋯O(3)viii | 2.11(3) | 2.967(2) | 173(3) |
| O(7w)–H(72w)⋯N(1)ix | 2.06(3) | 2.806(3) | 162(3) |
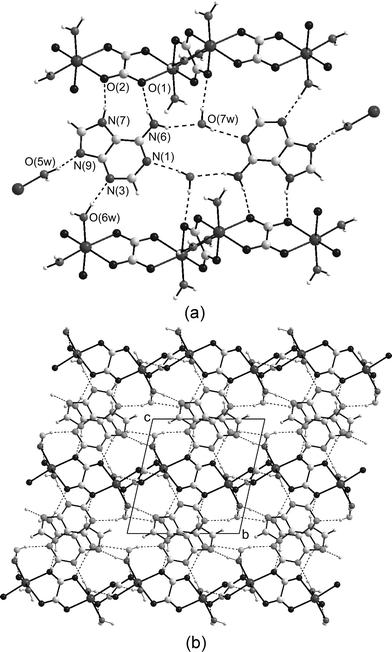 | ||
| Fig. 3 (a) View illustrating the hydrogen-bonded network (dashed lines) around the 7H-adenine tautomer. Out-of-plane O5w–H⋯O7w interactions have been omitted for the sake of the clarity. (b) Packing of the layers in the crystal building of compound 2 viewed along the crystallographic a-axis. | ||
Adenine rings of neighbouring aggregates are parallel stacked to afford wide layers of adenine and water molecules parallel to the crystallographic ab plane with a thickness of 7 Å. Polymeric metal-oxalato chains adhere to both sides of these wide adenine–water layers by means of a strong hydrogen bonding network involving all the potential donor/acceptor sites of the aggregates. The N6-H62 and N7-H7 groups of the adenine Hoogsteen face act as hydrogen bond donors to two oxygen atoms (O1 and O2) belonging to the ox1 oxalato bridge, giving an almost planar nine-membered ring. The N3 and N9 acceptor positions establish strong Ow–Hw⋯N hydrogen bonds with coordinated water molecules from adjacent chains placed in the same plane. The quite common distorted tetrahedral geometry around crystallization water molecules is achieved by out-of-plane contacts with the bound O5w water molecule from a neighbouring chain. The polymeric inorganic chains are further cross-linked by means of O6w–H61w⋯O4 (ox2) hydrogen bonds. So that, the crystal building of 2 could be alternatively described as a layered structure built up of sheets of neutral metal-oxalato chains along the crystallographic ab plane and adenine–water layers inserted among them. Each wide organic layer serves as “double-sided adhesive tape” to tightly join adjacent inorganic layers (Fig. 3).
The adenine nucleobase exists in the lattice in its 7H-tautomer form which has been unambiguously characterized by localization in the difference Fourier synthesis map of the proton attached to the N7 site, the aforementioned hydrogen bonding pattern, and the internal angle at N7 (C5–N7–C8: 106.5(2)°) which is significantly larger than that involving the deprotonated N9 position (C4–N9–C8: 103.9(2)°). It is known that the protonation at N sites in the adenine does not modify substantially the bond distances but it enlarges the endocyclic C–N–C angle up to ca. 4°.48 For comparison, these angles are 103.5 and 105.6° in the compound (9H-adenine)·2H2O.49
It is now well-documented that DNA bases can undergo proton shifts while keeping their neutrality and form different tautomers. Each tautomer has a specific H-bonding donor and acceptor pattern, which increases the possibility of mispairing of purine and pyrimidine bases in the DNA, leading to spontaneous point mutations in the genome.50 Due to its biological impact, the tautomerism of nucleobases has been largely studied using a variety of experimental and theoretical methods.8 Factors such as excitation, chemical modification, metallic cation interaction, electron attachment, and irradiation have been found to be responsible for the equilibrium between the most stable tautomer and the remaining forms of DNA bases. Additionally, hydration plays a significant role in the tautomeric processes and the water molecules may dramatically change the stability of different tautomeric forms of DNA bases through hydrogen bonding interactions, increasing the populations of the non-canonical tautomers. For adenine, a large number of studies have shown that the 9H-tautomer is energetically favored both in vacuo and water solution.51–55 The major 9H-adenine tautomer is of the order of 7–8 kcal mol−1 more stable than the minor 7H-adenine form in gas phase,51 but the inclusion of bulk electrostatic interactions (such as the interaction with polar solvent molecules) significantly stabilizes the 7H-adenine tautomer to approximately 2 kcal mol−1 above the 9H-tautomer,52 due to the much higher dipole moment of the 7H-adenine.53 A total dipole moment of 7.3 D has been computed for the adenine moiety in 2 [see ESI†]. This value is somewhat higher than those previously reported for optimized 7H-adenine tautomer in gas-phase (6.2–6.8 D)51–54 which substantially increase in water solution up to ∼10.2 D.55 For comparison, a dipole moment of 2.4 D has been obtained for the 9H-adenine tautomer in the solid state21 which matches those reported for this tautomeric form in vacuo (2.4–2.7 D)52–54 and it is slightly lower than the values reported in solution (∼3.2 D).55
Based upon these findings, previous studies have suggested that 7H-adenine tautomer might be present in a relatively large concentration in aqueous solution (of ca. 20%) and biological systems.56 Nevertheless, most of the known solid-state structures containing non-coordinated adenine molecules have H atoms at N9.21,47,49,57–59 To our knowledge, compound 2 represents the first X-ray crystallography characterization of the 7H-amino tautomer of the adenine nucleobase as free molecule (without metal coordination). Twofold N1,N7-protonated forms have been characterized for the compounds (1H,7H-ade)2(I3)2(I2)5(H2O)2,60 (1H,7H-ade)4(calix)·14H2O (calix = p-sulfonatocalix(4)arene),61 and (1H,7H-ade)2Cl[ZnCl3(1H,9H-ade-κN7)]·H2O62 but it is a cationic species rather than a tautomeric form because, strictly speaking, tautomers should be neutral.
The proton is only transferred to the N7 site of the neutral adenine in transition-metal complexes where the nucleobase behaves as either N9-monodentate or an N9,N3-bridging ligand and the resulting tautomer is commonly stabilized through a seven-membered ring maintained by means of simultaneous hydrogen bonds of the N7–H and N6–H groups to either chloride ion or water molecule as acceptor.25, 63 Similarly, the rare 1H-64 and 3H-amino65 tautomers have only been characterized as N9,N7-bidentate ligands. So that, the efficient stabilization of the 7H-adenine tautomer in the crystal structure of compound 2 seems to be clearly related to the molecular recognition between the nucleobase, the water molecules and the manganese-oxalato framework, because the proton transfer from N9 to N7 favors the formation of the N9⋯H51w–O5w interaction and the hydrogen-bonded ring involving the N7–H7⋯O2 (ox1) interaction. The first contact is the shortest among all interactions of this kind in the crystal packing of compound 2, and the structural parameters for the second one are within the lower range of values reported for hydrogen bonds involving the N7–H group of both neutral and cationic adenine moieties (N7⋯O: 2.617–3.060 Å, H7⋯O: 1.612–2.389 Å, N7–H7⋯O: 149–174°).15 Analogous hydrogen-bonded rings involving the adenine Hoogsteen face have been reported for the (adeninium)2[Co(H2O)2(7H-ade-κN9)2](SO4)2·6H2O66 and [Cu2(7H-ade-κN3,N9)4(H2O)2](ClO4)4·2H2O24 complexes, and the sulfate57 and nitrate58 salts of the 9H,1H,7H-adeninium dication.
Nine-membered rings formed by doubly hydrogen-bonded interactions of the Hoogsteen face are common in both artificial67 and biological systems, but most of them imply O/N–H⋯N7 interactions with the N7 site as H-bond acceptor. Hoogsteen AT base-parings found in different structures of DNA and RNA,68 and the Asn/Glu-adenine interaction in protein–nucleic acid complexes are sustained by this kind of hydrogen-bonded rings.69 Indeed, our first attempt to optimize the hydrogen bond environment around the 7H-adenine tautomer in 2 by using DFT calculations on a model containing a non-coordinated oxalato group led to a proton transfer from N7 to one oxygen atom of the oxalato to form a strong O–H⋯N7 hydrogen bond [O⋯N7: 2.835 Å, H⋯N7: 1.84 Å and ∠O–H⋯N7: 168°].
A more complete model with a dimeric [(H2O)2(ox)Mn(µ-ox)Mn(ox)(H2O)2] entity does not exhibit the transfer of the H7 atom and the structural parameters of the hydrogen-bonding network around the adenine moiety are quite similar to those found for compound 2 (see ESI†).
Several reports have suggested that the reduced stability of the N7-Hvs. the N9-H adenine tautomer is explained by the repulsion between lone pairs of the N3 and N9 and by the steric hindrance between the N7-H and the N6-amine.54 Indeed, optimized geometries of the 7H-adenine tautomer in the gaseous phase exhibit a partial sp3 pyramidalization of the amino group nitrogen atom with an out-of-plane conformation of their hydrogen atoms to avoid this steric hindrance; a conformation which is energetically unfavorable. Nevertheless, the adenine moiety in compound 2 and in the DFT optimized model obtained taking into account the hydrogen-bonding network around the tautomer show a significant decrease in the degree of pyramidality of the amino group which is accompanied with a substantial shortening of the C(6)–N(6) bond (from 1.383 Å for the isolated adenine up to 1.328 Å for 2). This indicates an essential strengthening of the conjugation between the π system of the purine bicyclic and the lone pair of the nitrogen atom. Therefore, one can assume that a decrease in the pyramidality of the amino group is forced by the intermolecular hydrogen bonds established by the 7H-adenine tautomer with the water molecules and the manganese-oxalato framework.
| Cu(1a)–O(1a) | 1.929(5) | Cu(1b)–O(1b) | 1.936(6) |
| Cu(1a)–O(2a) | 1.903(6) | Cu(1b)–O(2b) | 1.919(5) |
| Cu(1a)–N(9a) | 1.971(5) | Cu(1b)–N(9b) | 1.969(6) |
| Cu(1a)–O(5wa) | 1.916(6) | Cu(1b)–O(5wb) | 1.961(6) |
| Cu(1a)–O(1a)i | 2.848(6) | Cu(1b)–O(1b)i | 2.844(7) |
| Cu(1b)–O(5wb)ii | 2.712(6) |
| D–H⋯Ab | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|
| a Symmetry codes: (i): 1 + x, y, z; (ii) −1 + x, y, z; (iii) 2 − x, 1 − y, 1 − z; (iv) 1 + x, 1 + y, z; (v) −x, 1 − y, −z; (vi) −1 + x, −1 + y, z. b D: donor, A: acceptor. | |||
| N(6a)–H(61a)⋯O(2b)iii | 2.04 | 2.888(9) | 169 |
| N(6a)–H(62a)⋯O(3b)i | 2.00 | 2.821(9) | 161 |
| N(7a)–H(7a)⋯O(3b)i | 2.12 | 2.877(9) | 147 |
| N(6b)–H(61b)⋯O(1a)iv | 2.07 | 2.926(9) | 178 |
| N(6b)–H(62b)⋯O(4a)v | 2.00 | 2.845(9) | 169 |
| N(7b)–H(7b)⋯O(4a)v | 2.07 | 2.841(9) | 150 |
| O(5wa)–H(51wa)⋯N(1b)vi | 1.89 | 2.720(9) | 163 |
| O(5wa)–H(52wa)⋯N(3a) | 1.94 | 2.636(9) | 134 |
| O(5wb)–H(51wb)⋯N(1a)iii | 1.89 | 2.739(9) | 171 |
| O(5wb)–H(52wb)⋯N(3b) | 1.96 | 2.695(9) | 140 |
 | ||
| Fig. 4 ORTEP depiction of the complex units of the compound 3 (50% probability thermal ellipsoids). | ||
In both complex fragments, the Cu(II) atoms are coordinated to two oxygen atoms from a bidentate oxalato ligand, one water molecule and the imidazole N9 atom from the adenine ligand with Cu1–N9 bond distances of 1.971(5) Å (A) and 1.969(6) Å (B). The binding of metal ions to the most basic N9 donor atom of the adenine nucleobase (pKa = 9.8)70 is largely supported from a structural point of view as could be found in a CSD search for nearly 40 compounds, and the shift of the hydrogen atom to the pyrimidinic N1 atom or the major groove N7 position is observed for neutral ligands. This fact is in agreement with the relative basicity of the N-rich adenine nucleobase (N9 > N1 > N7 > N3 > N6-exocyclic).2 The CuO3N chromophores are essentially coplanar (maximum atomic deviation 0.08 Å for O1a) with copper atoms being displaced by 0.04 Å (Cu1a) and 0.06 Å (Cu1b) from the least-square plane. The adenine and oxalato ligands make dihedral angles of 8.8° (A) and 11.3° (B), respectively. Each complex unit is connected to two symmetric related ones through weaker axial Cu–O interactions to create neutral ribbons with Cu⋯Cu distances coincident with the a parameter of the unit cell [3.587(1) Å]. Whereas the metallic atoms of the A chain are only bridged by the coordinated O1 oxygen atom of the oxalato ligand [Cu(1a)–O(1a)i: 2.848(6) Å], the copper centres in the B chain are additionally joined by the bound water oxygen atom [Cu(1b)–O(1b)i: 2.844(7) Å, Cu(1b)–O(5wb)ii: 2.712(6) Å]. Taking into account all the axial Cu–O interactions, the coordination geometry of Cu(1a) may be described as square pyramidal, and that of Cu(1b) as elongated octahedral. The axial Cu–Oox bond distances exceed the equatorial ones by ca. 0.95 Å, and are similar to those reported in analogous oxalato-copper(II) complexes where the C2O42− group acts in a bidentate-monodentate bridging mode (µ-1,1,2-oxalato).71 In both chains, all the adenine ligands are located in the same side of the polymeric framework and they are ring-to-ring stacked, suggesting that π–π stacking interactions contribute to the formation of the one-dimensional chains. The planarity of the complex entities is mainly sustained by an intramolecular hydrogen bond involving the coordinated water molecule (as donor) and the N3 site of the nucleobase (as acceptor) but also the parallel stacking of the complex entities presumably play an important role. Indeed, DFT calculations on an isolated complex unit gave an optimized model which reproduces with acceptable precision the experimental bond distances and angles, and the structural parameters of the intramolecular hydrogen bond, but the O3N donor set shows a significant tetrahedral distortion with a N9–Cu–O bond angle of 158.9°, and the overall structure is not nearly planar with a dihedral angle between the oxalato and adenine ligand of 26.0°.
Polymeric one-dimensional chains are further interlinked through an intricate hydrogen bonding network spreading out along the bc plane. Watson–Crick-like hydrogen bonds are observed between the adenine moiety and the coordinated oxygen atoms from the oxalato and aqua ligands belonging to the adjacent complex unit. The Hoogsteen donor sites of the nucleobase (N7H and N6H groups) are hydrogen-bonded to one noncoordinated oxygen atom from one oxalato ligand belonging to another adjacent complex entity. The supramolecular assembling of the structural units driven by the above-described combination of coordinative linkages and hydrogen bonding interactions creates a 3D porous structure72 showing two distinct spherical channels along the a axis of the unit cell with diameters of about 9 and 7 Å, respectively (Fig. 5). The first one is located in the ab face of the unit cell and its inner walls are occupied by the free oxalato oxygen atoms, whereas the walls of the second one (placed in the ac faces of the unit cell) are formed by the C2–H2 bonds of the pyrimidinic rings. Calculations using PLATON33 showed that the effective volume for inclusion is about 194 Å3 cell−1 comprising 17% of the crystal volume. These parallel cavities are filled by guest water molecules with a high crystallographic disorder.
 | ||
| Fig. 5 View along the a axis of the arrangement of the polymeric chains in 3 showing the hydrogen bonds between them. Cavities are occupied by disordered water guest molecules, which have been removed for clarity. | ||
CCDC reference numbers 278470–278472.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b510018f
Magnetic measurements
The thermal evolution of the magnetic molar susceptibility (χM), with maximum values at 13 (1) and 11 K (2), and the temperature dependence of the product χMT of compounds 1 and 2 (Fig. 6) are indicative of antiferromagnetic coupling between the manganese(II) centres through the bridging oxalato ligands. The best fits of the experimental magnetic data by means of a S = 5/2 Fisher antiferromagnetic chain model (H = − J∑SiSi + 1)73 gave J = −1.99 cm−1, g = 2.00 for 1, and J = −1.82 cm−1, g = 2.00 for 2. The replacement of oxygen by nitrogen atoms in the manganese(II) coordination polyhedron (MnO6 and MnO5N chromophore for 2 and 1, respectively) is accompanied by an increase in the antiferromagnetic coupling due to the smaller electronegativity of the donor atom, which interacts more strongly with the manganese d orbitals and induces a strong hybridization toward the bridging ligand, thus favoring a larger bridge orbital overlap. Indeed, a J value of −2.48 cm−1 has been reported for the dinuclear [Mn2(µ-ox)(bispictn)2](ClO4)2 compound with a O2N4 donor set.39 A similar dependence of the magnitude of the exchange coupling with the nature of the donor atoms around the metal centre has been previously reported by some of us for oxalato-bridged nickel(II) complexes.74 | ||
| Fig. 6 Thermal dependence of χMT (△) and χM (○) curves for compounds 1 (up) and 2 (bottom). (–) best theoretical fit (see text). | ||
For compound 3, the variation of the inverse susceptibility (χM−1) is well described in all studied temperature ranges by the Curie–Weiss law with a θ value of −0.6 K (Fig. 7). This fact and the temperature dependence of the χMT product (χM is the magnetic susceptibility per copper atom) are indicative of the presence of very weak antiferromagnetic interactions between the metal centres, according to the most important exchange pathways in 3 which involve the long axial Cu–O bonds.71
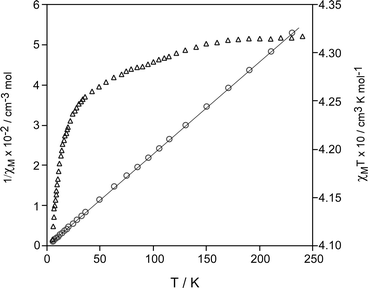 | ||
| Fig. 7 Plots of the thermal dependence of χMT (△) and 1/χM (○) for compound 3. | ||
Acknowledgements
This work was supported by the Ministerio de Ciencia y Tecnología (MAT2005-03047) and the Universidad del País Vasco/Euskal Herriko Unibertsitatea (9/UPV 00169.310-15329/2003). J. P. G.-T. thanks Eusko Jaurlaritza/Gobierno Vasco for a predoctoral fellowship.References
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737 CAS; F. Crick, Nature, 1974, 248, 766 CrossRef CAS.
- B. Lippert, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley and Sons, New York, 2005, vol. 54, p. 385-447 Search PubMed; B. Lippert, Coord. Chem. Rev., 2000, 200–202, 487 Search PubMed.
- B. Lippert, J. Chem. Soc., Dalton Trans., 1997, 3971 RSC.
- S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9 RSC; J. A. R. Navarro and B. Lippert, Coord. Chem. Rev., 2001, 222, 219 CrossRef CAS.
- E. Bugella-Altamirano, D. Choquesillo-Lazarte, J. M. González-Pérez, M. J. Sánchez-Moreno, R. Marín-Sánchez, J. D. Martín-Ramos, B. Covelo, R. Carballo, A. Castiñeiras and J. Niclós-Gutiérrez, Inorg. Chim. Acta, 2002, 339, 160 CrossRef CAS; A. Marzotto, D. A. Clemente, A. Ciccarese and G. Valle, J. Crystallogr. Spectrosc. Res., 1993, 23, 119 CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1211; A. C. Chen and A. D. Frankel, J. Am. Chem. Soc., 2004, 126, 434 CrossRef CAS.
- J. Anastassaopoulou, J. Mol. Struct., 2003, 651–653, 19 CrossRef CAS.
- J. Sponer, J. Leszczynski and P. Hobza, Biopolymers, 2002, 61, 3 CrossRef CAS.
- J. Sponer, J. Leszczynski and P. Hobza, J. Mol. Struct., 2001, 573, 43–53 CrossRef CAS.
- V. J. Derose, S. Burns, N.-K. Kim and M. Vogt, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, University of Bern, Switzerland, 2004, vol. 8, p. 787-813 Search PubMed.
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215 RSC.
- R. H. Fish and G. Jaouen, Organometallics, 2003, 22, 2166 CrossRef CAS and references therein.
- H. Sigel, Chem. Soc. Rev., 2004, 33, 191 RSC; S. J. Berners-Price and P. J. Sadler, Coord. Chem. Rev., 1996, 151, 1 CrossRef CAS.
- M. Vaduz and K. Aoki, Inorg. Chim. Acta, 2000, 311, 15 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B, 2002, 58, 380 CrossRef.
- P. I. Vestues and E. Sletten, Inorg. Chim. Acta, 1981, 52, 269 CrossRef CAS.
- E. Sletten, J. Sletten and N. A. Froystein, Acta Chem. Scand., Ser. A, 1988, 42, 413 Search PubMed.
- H. L. Laity and M. R. Taylor, Acta Crystallogr., Sect. C, 1995, 51, 1791 CrossRef; W. S. Sheldrick, Z. Naturforsch., B, 1982, 37, 653.
- S. Das, C. Madhavaiah, S. Verma and P. K. Bharadwaj, Inorg. Chim. Acta, 2005, 358, 3236 CrossRef CAS; M. Quirós-Olazabal, J. M. Salas-Peregrín, M. P. Sánchez and R. Faure, An. Quim., 1990, 86, 518.
- J. P. García-Terán, O. Castillo, A. Luque, U. García-Couceiro, P. Román and L. Lezama, Inorg. Chem., 2004, 43, 4549 CrossRef CAS.
- J. P. García-Terán, O. Castillo, A. Luque, U. García-Couceiro, P. Román and F. Lloret, Inorg. Chem., 2004, 43, 5761 CrossRef CAS.
- P. de Meester, D. M. L. Goodgame, T. J. Jones and A. C. Skapski, Biochem. J., 1974, 139, 791 CAS; C. G. Hoogstraten, C. V. Grant, T. E. Horton, V. J. DeRose and R. D. Britt, J. Am. Chem. Soc., 2002, 124, 834 CrossRef CAS.
- M. V. Capparelli, D. M. L. Goodgame, P. B. Hayman and A. C. Skapski, Inorg. Chim. Acta, 1986, 125, L47 CrossRef CAS.
- A. Terzis, A. L. Beauchamp and R. Rivest, Inorg. Chem., 1973, 12, 1166 CrossRef CAS.
- P. De Meester and A. C. Skapski, J. Chem. Soc., Dalton Trans., 1972, 2400 RSC; A. C. Morel, D. Choquesillo-Lazarte, C. Alarcón-Payer, J. M. González-Pérez, A. Castañeiras and J. Niclós-Gutiérrez, Inorg. Chem. Commun., 2003, 6, 1354 CrossRef CAS.
- S. Kirschner, in Inorganic Synthesis, ed. E. G. Rochow, McGraw-Hill Book Co., New York, 1960, vol. 4 Search PubMed; H. Remy, in Treatise on Inorganic Chemistry, VCH, Weinheim, Germany, 1956 Search PubMed.
- A. Earnshaw, in Introduction to Magnetochemistry, Academic Press, London, 1968 Search PubMed.
- CrysAlis RED. Version 1.170, Oxford Diffraction, Wroclaw, Poland, 2003 Search PubMed.
- A. Altomare, M. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- G. M. Sheldrick, SHELXL97, Universitat of Göttingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, WINGX, A Windows Program for Crystal Structure Analysis, University of Glasgow, UK, 1998 Search PubMed.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. A, 1990, 46, C34.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 2000.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian 03, Revision B.04, Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed.
- S. Simizu and S. A. Friedberg, J. Appl. Phys., 1988, 63, 3557 CrossRef CAS.
- U. García-Couceiro, D. Olea, O. Castillo, A. Luque, P. Román, P. J. de Pablo, J. Gómez-Herrero and F. Zamora, Inorg. Chem., 2005, 44, 8343 CrossRef CAS; X. Fu, C. Wang and M. Li, Acta Crystallogr., Sect. E, 2005, 61, m1348; L. H. Zhu, M. H. Zeng and S. W. Ng, Acta Crystallogr., Sect. E, 2005, 61, m916; D. Deguenon, G. Bernardinelli, J. P. Tuchagues and P. Castan, Inorg. Chem., 1990, 29, 3031 CrossRef CAS.
- J. Glerup, P. A. Goodson, D. J. Hodgson and K. Michelsen, Inorg. Chem., 1995, 34, 6255 CrossRef CAS.
- H. Shi and P. B. Moore, RNA, 2000, 6, 1091 CrossRef CAS.
- K. Junean, E. Podell, D. J. Harrington and T. R. Cech, Structure, 2001, 9, 221 CrossRef.
- W. G. Scott, J. B. Murray, J. R. P. Arnold, B. L. Stoodard and A. Klug, Science, 1996, 274, 2065 CrossRef CAS.
- A. L. Feig, Met. Ions Biol. Syst., 2000, 37, 157 CAS.
- K. J. Baeyens, H. L. de Bondt, A. Pardi and S. R. Holbrook, Proc. Natl. Acad. Sci. USA, 1996, 93, 12851 CrossRef CAS.
- H. W. Pley, K. M. Flaverty and D. B. McKay, Nature, 1994, 372, 68 CrossRef CAS; W. G. Scott, J. B. Murray, J. R. P. Arnold, B. L. Stoddard and A. Klug, Science, 1996, 274, 2065 CrossRef CAS; L. M. Hunsicker and V. J. deRose, J. Inorg. Biochem., 2000, 80, 271 CrossRef CAS; N. Kisseleva, A. Khvorova, E. Westhof and O. Schiemann, RNA, 2005, 11, 1 CrossRef CAS.
- G. de Munno, M. Medaglia, D. Armentano, J. Anastassopoulou and T. Theophanides, J. Chem. Soc., Dalton Trans., 2000, 1625 RSC; C. V. Grant, V. Frydman and L. Frydman, J. Am. Chem. Soc., 2000, 122, 11743 CrossRef CAS; M. Sabat, R. Cini, T. Haromy and M. Sundaralingham, Biochemistry, 1985, 24, 7827 CrossRef CAS; K. Aoki, J. Chem. Soc., Chem. Commun., 1976, 748 RSC.
- D. Dobrzynska and L. B. Jerzykiewicz, J. Am. Chem. Soc., 2004, 126, 11118 CrossRef CAS.
- A. Marzotto, A. Ciccarese, D. A. Clemente and G. Valle, J. Chem. Soc., Dalton Trans., 1995, 1461 RSC.
- S. M. Tretyak, V. V. Mitkevich and L. F. Sukhodub, Crystallogr. Rep., 1987, 32, 1268 CAS.
- M. F. Goodman, Nature, 1995, 378, 237 CrossRef CAS.
- Y. Huang and H. Kenttamma, J. Phys. Chem. A, 2004, 108, 4485 CrossRef CAS and references therein.
- J. Gu and J. Leszczynski, J. Phys. Chem. A, 1999, 103, 2744 CrossRef CAS.
- S. P. Perun, A. L. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2005, 127, 6257 CrossRef CAS.
- L. M. Salter and G. M. Chaban, J. Phys. Chem. A, 2002, 106, 4251 CrossRef CAS; A. L. Laxer, D. T. Mayor, H. E. Gottlied and B. Fischer, J. Org. Chem., 2001, 66, 5463 CrossRef CAS.
- B. Mennucci, A. Toniolo and J. Tomasi, J. Phys. Chem. A, 2001, 105, 4749 CrossRef.
- C. E. Crespo-Hernández, B. Cohen, P. M. Hare and B. Holher, Chem. Rev., 2004, 104, 1977 CrossRef CAS.
- V. Langer and K. Huml, Acta Crystallogr., Sect. A, 1984, 40, C86.
- G. L. Hardgrove, J. R. Einstein, B. E. Hingerty and C. H. Hei, Acta Crystallogr., Sect. C, 1983, 39, 88 CrossRef.
- C. M. Weeks, D. C. Rohrer and W. L. Duax, Science, 1975, 190, 1096 CrossRef CAS; M. A. Serra, B. K. Dorner and M. E. Silver, Acta Crystallogr., Sect. C, 1992, 48, 1957 CrossRef.
- Z. Wang, Y. Cheng, C. Liao and C. Yan, CrystEngComm., 2001, 50, 1 RSC.
- J. L. Atwood, L. J. Barbour, E. S. Dawson, P. C. Junk and J. Kienzle, Supramol. Chem., 1996, 7, 271 CrossRef CAS.
- M. R. Taylor and J. A. Westphalen, Acta Crystallogr., Sect. A, 1981, 37, C63.
- P. de Meester and A. C. Skapski, J. Chem. Soc. A, 1971, 2167 RSC; T. Suzuki, Y. Hirai, H. Monjushiro and S. Kaizaki, Inorg. Chem., 2004, 43, 6435 CrossRef CAS.
- W. S. Sheldrick, H. S. Hagen-Eckhard and S. Hebb, Inorg. Chim. Acta, 1993, 206, 15 CrossRef CAS.
- P. X. Rojas-González, A. Castiñeiras, J. M. González-Pérez, D. Choquesillo-Lazarte and J. Niclós-Gutiérrez, Inorg. Chem., 2002, 41, 6190 CrossRef CAS.
- P. de Meester and A. C. Skapski, J. Chem. Soc., Dalton Trans., 1973, 1596 RSC.
- V. Langer, K. Huml and J. Zachova, Acta Crystallogr., Sect. B, 1979, 35, 1148 CrossRef; J. Maixner, J. Zachova and K. Huml, Collect. Czech. Chem. Commun., 1993, 58, 861 CrossRef CAS; Z. Travnicek, J. Marek, K. Dolezal and M. Strnad, Z. Kristallogr., 1997, 212, 538 CAS.
- E. Cubero, N. G. A. Abrescia, J. A. Subirane, F. J. Luque and M. Orozco, J. Am. Chem. Soc., 2003, 125, 14603 CrossRef CAS.
- A. C. Cheng and A. D. Frankel, J. Am. Chem. Soc., 2004, 126, 434 CrossRef CAS.
- M. M. Taqui-Khan and C. K. Krishnamoorthy, J. Inorg. Nucl. Chem., 1971, 42, 1417 CrossRef.
- H. Nuñez, J. J. Timor, J. Server-Carrió, L. Soto and E. Escrivá, Inorg. Chim. Acta, 2001, 318, 8 CrossRef CAS and references therein.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC.
- M. E. Fisher, Am. J. Phys., 1964, 32, 343.
- P. Román, C. Guzmán-Miralles, A. Luque, J. I. Beitia, J. Cano, F. Lloret, M. Julve and S. Alvarez, Inorg. Chem., 1996, 35, 3741 CrossRef CAS; O. Castillo, A. Luque, P. Román, F. Lloret and M. Julve, Inorg. Chem., 2001, 40, 5526 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Binding patterns of the adenine and purine bases reported for transition metal coordination compounds compiled in the CSD database. Usual protonation sites of the adenine and purine bases. Selected bond angles (°) in compounds 1–3. Structural parameters for the DFT optimized structures of the isolated 7H-adenine tautomer and the model with the hydrogen-bonding environment found in compound 2. Views of the crystal packing of compound 3. Thermoanalytical (TG/DTA) curves for compounds 1–3. See DOI: 10.1039/b510018f |
| This journal is © The Royal Society of Chemistry 2006 |