Metal complex SERMs (selective oestrogen receptor modulators). The influence of different metal units on breast cancer cell antiproliferative effects
Anne
Vessières
a,
Siden
Top
a,
Wolfgang
Beck
b,
Elizabeth
Hillard
a and
Gérard
Jaouen
*ab
aLaboratoire de Chimie et Biochimie des Complexes Molèculaires, UMR CNRS 7576, Ecole Nationale Supèrieure de Chimie de Paris, 11 rue Pierre et Marie Curie, 75231, Paris cedex 05, France. E-mail: gerard-jaouen@enscp.fr; Fax: 01 43 26 00 61; Tel: 01 43 26 95 55
bDepartment of Chemistry, Universität München, Butenandtstr. 5-13, 81377, München, Germany
First published on 27th September 2005
Abstract
The selective oestrogen receptor modulator tamoxifen is a leading agent in the adjuvant treatment of breast cancer. Several organometallic moieties have been vectorised with tamoxifen, in order to improve on the latter's antiproliferative properties by the addition of a potentially cytotoxic moiety, and have been evaluated versus both oestrogen receptor positive (MCF7) and oestrogen receptor negative (MDA-MB231) breast cancer cells. For tamoxifen analogues with ((R,R)-trans-1,2-diaminocyclohexane)platinum(II), cyclopentadienyl rhenium tricarbonyl, and ruthenocene tethers, there was no enhancement of the antiproliferative effect on oestrogen receptor positive cells, nor any cytotoxic effect on oestrogen receptor negative cells, while those containing cyclopentadienyl titanium dichloride showed an oestrogenic effect. However, compounds where ferrocene replaces tamoxifen's phenyl ring were strongly cytotoxic against both cell lines. The synthesis and biological results of these compounds is reviewed and placed in the historic context of inorganic compounds in therapy.
 Elizabeth Hillard Elizabeth Hillard | Elizabeth Hillard was born in 1971 in Ripon, Wisconsin. She received a BA in Theatre from the University of Alaska in Fairbanks in 1994, and has worked for several years for various production companies. She returned to the University to obtain a BS in Chemistry in 1999, and did undergraduate research at University of Southern California in Summer of 1998. She then joined Professor Al Cotton's research group at Texas A & M University, and received her PhD in Spring of 2003. In the fall of 2003, she joined Professor Gèrard Jaouen's group at Ecole Nationale Superieure de Chimie de Paris with the assistance of a National Science Foundation postdoctoral fellowship. |
 Gérard Jaouen Gérard Jaouen | Gèrard Jaouen was born in 1944 in France. He did graduate work at the University of Rennes, submitting his doctoral thesis in organic chemistry in 1969 and earning a Doctorat d'Etat in Physical Sciences in 1973 in the laboratory of Professor R. Dabard. He spent the year 1973–1974 at Cambridge working with Professor (now Lord) Jack Lewis. He became a member of the CNRS in 1970, was appointed Directeur de Recherche in 1976, and became Professor at the Ecole Nationale Supèrieure de Chimie in Paris in 1983, where in 1984 he set up a CNRS Associated Research Unit. His research interested first centered on establishing a series of new concepts in the use of organometallics in organic synthesis, particularly in the arene chromium tricarbonyl series. This work takes advantage of the differences in reactivity between free and complexed aromatics in synthesis, including organometallic synthesis of chiral molecules. At the time this theory was in its infancy and had not reached the level of development it has today, and his groundbreaking studies are now found in most organometallic chemistry textbooks. After some work on asymmetric metals (chromium) and reaction mechanism (metallic hydrides), Dr Jaouen decided to orient his interest in a new direction: that of bioorganometallics, which offers a vast potential for organometallic chemistry. This field appears rich in promise and these novel ideas have been presented in lectures all over the world. Professor Jaouen is the author of 300 papers and 14 reviews, and holds 10 patents. He has been elected to the “Institut Universitaire de France” in 1997, in recognition of the very high level of originality and creativity in pioneering this new area of bioorganometallic chemistry, one of the beautiful examples of interdisciplinary science. In 2001 he received in 2001 a von Humboldt award in Berlin, was elected to the Royal Society of Chemistry as the Centenary Lecturer 2002–2003 and was the recipient of the Pioneer Award (AIC) in Boston 2002, and of the Bioorganometallic Award in Zurich 2004. |
Introduction
The social and medical impact of molecules which have been recognised for their oestrogenic or antioestrogenic properties impinges on a variety of fields such as contraception,1 osteoporosis,2 menopause,3 hormone dependent cancers,4 and possibly Alzheimer's5 and coronary disease.6 It is particularly in the field of breast cancer, which touches one woman in eight in the West,7 where the contribution of tamoxifen (TAM) has been especially positive.8 This molecule, shown in Chart 1 as its active metabolite, hydroxytamoxifen (OH-TAM), has significantly contributed to the current (US) patient survival rate of 88%.9 Due to its efficacy against breast cancer and high tolerance among patients, all while promoting desirable effects such as the maintenance of bone density;10 tamoxifen to date remains the SERM (Selective Oestrogen Receptor Modulator) prototype, while estradiol (E2), a natural molecule, is the oestrogen of reference.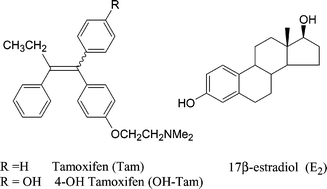
Because the potency of tamoxifen arises from its antioestrogenicity in the breast, patients diagnosed with hormone dependent breast cancer, where the oestrogen receptor is present (ER+), have a more positive prognosis than those with hormone independent cancers, which account for approximately 1/3 of cases.11 Nonetheless, even in ER+ cancers, long-term tamoxifen therapy (over five years) frequently leads to drug resistance.12 The situation has been complicated by the discovery in 1996 of a second oestrogen receptor, ERβ,13 so named to differentiate from the ER now called ERα,14 which was first cloned in the 1980's. Malignant breast tissue may express ERα alone, a combination of ERα and ERβ, or no ER, and in rare cases ERβ alone.15 ERβ has also been detected in some breast cancer cell lines,16 notably MDA-MB231, the standard ERα-negative (ER−) cell line. Due to a moderate LBD (ligand binding domain) homology (about 60%),13b,17 many SERMs bind as readily to ERβ as to ERα,18 yet the detection of ERβ in breast cancer tumours indicates a poor prognosis for tamoxifen treatment,15d suggesting that the mechanism of action for the two receptors is not identical. The search for new SERMs able to clearly differentiate the two oestrogen receptors is one approach to increasing breast cancer survival.19 Another approach is that of combination therapy, where SERMs and toxic chemotherapeutic agents are simultaneously administered in order to quickly kill malignant cells before the onset of drug resistance.20 An alternative would be the design of a single drug which combines cytotoxic and antioestrogenic properties, and the contribution of transition metal complexes in the search for such active molecules is of particular interest.
Brief outline of the mechanism of action of SERMs
Although tamoxifen has been quite successful in the treatment of breast cancer, it has the unwelcome side effect of increasing the occurrence of uterine cancer.21 This flaw arises from the versatility of the oestrogen receptor in transcription modulation, a process which depends ultimately on whether activator or repressor proteins are recruited to the ER. Coactivator recruitment is now known to be controlled on at least three levels: (a) the conformation of the receptor in response to the structure of the bioligand; (b) the mechanism of dimer activation of the receptor with DNA; and (c) the levels of coregulators present in the target bodies.22 Adding to the complexity, it is worthwhile to note that a great number of genes are under the influence of the ER, whose role in breast cancer is not completely understood.23Crystal structures of ERα and ERβ with estradiol, tamoxifen and raloxifene24 have shown that the nature of the bioligand causes a change in the conformation of the receiving protein. Although the wide variety of experiments tends to make the situation rather complicated, two extreme cases illustrate the action of tamoxifen versus that of estradiol. After the capture of estradiol by the receptor, there is a snapping shut of a “mouse trap” with helix 12 closing onto helix 4. This closed conformation is stabilised by saline bridges, and positions helix 12 in such way that it can join certain coactivators if they are present in the cellular environment. On the other hand, when OH-TAM is associated with the receptor LBD, the steric effect of the dimethylamino side chain (O(CH2)2NMe2) is crucial. It opens a pocket in a flexible part of the protein, preventing the folding up of helix 12 onto helix 4, a conformation which seems to prevent coactivator binding in the breast. This open form is stabilised by association of the amine with the residue Asp 351 of the receptor. The selection of the coregulators present in the environment thus depends on the conformation of the bioligand/receptor complex.
Transcription can also be regulated by the manner in which the ER binds with DNA. It may do so through direct association with oestrogen response elements of DNA (ERE pathway),25 or in an indirect fashion by association with activator proteins which are themselves associated with another region of the DNA, (the AP1 pathway). The ERE and AP1 pathways each submit different genes to ER control. The ER bound to E2 or TAM may associate with DNA through either pathway, although ERβ has been found to function solely through the ERE site when bound to E2.26 The ER binding site also seems to influence whether coactivators or corepressors are recruited. For example, in the uterus, tamoxifen recruits corepressors to the genes under ERE control, and coactivators to those under AP-1 control.
The availability of certain coregulators in a given organ tissue is also responsible for agonist or antagonist behaviour.27 For example, E2 is an agonist in both the breast and uterus, and promotes expression of genes under control of both ERE and AP1 pathways due to the recruitment of a variety of coactivators. Tamoxifen, on the other hand, is an antagonist in breast cancer cells on the level of both the ERE and AP1 pathways due to the recruitment of certain corepressors. Interestingly, tamoxifen-bound ERβ acts as a promoter of gene expression through the AP1 pathway, where this isoform recruits coactivators.28 AP1 controlled genes such as c-Myc and IGFI have been implicated in uterine cancer, and thus tamoxifen acts like oestrogen in the uterus. The high availability of the coactivator SRC-1 in the uterus, as opposed to the breast may explain this phenomenon. These various effects, namely nature of the SERM and the ER, the type of organ, and the selectivity of genes brought into play provide a framework for the development of SERMs with optimised selectivity.
The use of tamoxifens as vectors for metal systems
The toxic molecules used in chemotherapy, as opposed to endocrine therapy, are often undermined by their non-specific character which can cause harm to both healthy tissue and cancerous target cells, resulting in many undesirable side effects. Because of its recognition for the ER, the use of tamoxifen as an active conveyor of potentially cytotoxic metal moieties should therefore theoretically solve this selectivity problem. Ideally, the antioestrogenic effects of the vector should be conserved along with the addition of the intrinsic cytotoxic function of the metal complex, and this dual-function drug should be effective against both tamoxifen-responsive and -resistant tumours. It should be mentioned that even tamoxifen-resistant tumours may possess ER, and thus the potential for selective cytotoxicity is especially important in these cases. This report reviews several metal-tamoxifen hybrids which have been synthesised and evaluated in our laboratory. We concentrate on the examples where both the products and their antiproliferative activity in vitro have been characterised. In many cases, success has been quite elusive, and the various counterproductive situations that we have encountered have taught us that this idea requires some adjustments.There are certain practical prerequisites that must be met in order for a candidate molecule to be efficacious against breast cancer cells, whether in vitro or in vivo. The compound must be lipophilic enough to pass through the phospholipid cell membrane. Lipophilicity is usually evaluated by the octanol/water partition coefficient, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w, and this value should be around 3.3 for the molecule to be expected to reach the cytoplasm. A second important consideration is the aptitude of the candidate to recognise the ER; this is expressed as the relative binding affinity (RBA) for the ER. RBA values of 10% and higher are generally considered promising. For example, among the inorganic complexes of the triphenylethylene Pt(II) type, the derivatives depicted in Chart 2 are characterised by a fast and effective synthesis. However, the relative binding affinity (RBA) for these compounds is very low, approximately 1.3%.29 While the cytotoxicity of some of these compounds was satisfactory, with IC50 values on the order of 1 μM, these drugs do not confer any particular advantage over cisplatin due to their lack of specificity.
Po/w, and this value should be around 3.3 for the molecule to be expected to reach the cytoplasm. A second important consideration is the aptitude of the candidate to recognise the ER; this is expressed as the relative binding affinity (RBA) for the ER. RBA values of 10% and higher are generally considered promising. For example, among the inorganic complexes of the triphenylethylene Pt(II) type, the derivatives depicted in Chart 2 are characterised by a fast and effective synthesis. However, the relative binding affinity (RBA) for these compounds is very low, approximately 1.3%.29 While the cytotoxicity of some of these compounds was satisfactory, with IC50 values on the order of 1 μM, these drugs do not confer any particular advantage over cisplatin due to their lack of specificity.
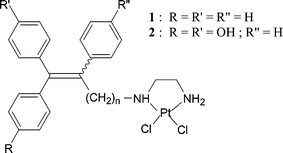 | ||
| Chart 2 Pt(II) Triphenylethylene derivatives. | ||
In the case of radiopharmaceutical metals, let us note the synthesis of chelates such as that appearing in Chart 3. The biological evaluation in vitro as well as in vivo showed that both the Tc and Re chelates derived from tamoxifen had a very limited association with respect to the ER, making them practically unusable as tumour imaging reagents (RBA <0.009%).30
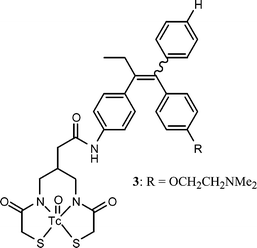 | ||
| Chart 3 | ||
With the aim of coupling tamoxifen to a boron neutron capture therapy (BNCT) agent, the boroxifen, with a nido carborane, was prepared, Chart 4.31 This compound, however, has no affinity for the oestrogen receptor due to the absence of the hydrogen bonding phenol group.32 Let us note that carboranes can be powerful agonists of oestrogen receptors.33
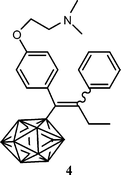 | ||
| Chart 4 Carborane-tamoxifen derivative: polyhedron represents HC2B9H10. | ||
These few examples show that, when coupling a metal functionality to a vector, the first requirement of suitable recognition for the target is not always reached. In the following examples receptor affinity is adequate, but even so, one can observe the diversity of behaviour of the metal complexes brought into play.
To solve these synthetic problems, as well as those associated with the question of resistance, undesirable side effects, and the narrow spectrum of application; researchers have synthesised a very great number of cisplatin analogues.37 Among the thousands of new molecules prepared, approximately only thirty have succeeded in reaching the clinical testing stage,38 of which only a half dozen have received approval for clinical use.39 Two molecules at this moment are routinely used: carboplatin [cis-diamine(1,1′-cyclobutanedicarboxylate)platinum(II)]40 and oxaliplatin [(trans-R,R-1,2-diaminocyclohexaneoxalo)platinum(II)],41 shown in Chart 5. Carboplatin is clearly an improvement over cisplatin for some tumours, with a similar activity but a lowered systemic toxicity.42 Oxaliplatin, in which the fragment (DACH)Pt, [(trans-R,R-1,2-diaminocyclohexane)platinum], is coordinated to an oxalate ligand, is commercially available under the names of Eloxtine, Dacplat or Dacotin. It acts as an effective third generation drug against certain cisplatin-resistant cancers such as colorectal cancer.43
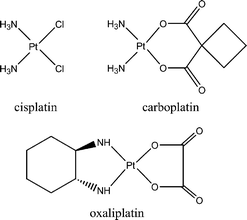 | ||
| Chart 5 Platinum drugs currently in clinical use. | ||
Biochemical tests have shown a moderate antiproliferative activity for oxaliplatin on the hormone dependent MCF7 breast cancer cell line, with an IC50 value of 7.4 μM.44 The antiproliferative action of oxaliplatin (as well as cisplatin and carboplatin) does not arise from antioestrogenicity, but from formation of DNA adducts leading to cell death. On this basis, we decided that the selectivity of oxaliplatin may be enhanced by tamoxifen vectorisation.45 The molecules shown in Chart 6 were synthesised via a McMurry coupling reaction, detailed in Scheme 1.46 The coupling reaction is very stereoselective in the case of R = H where the Z isomer is exclusively obtained, while in the case of R = OH, the reaction yielded an E/Z ratio of 80/20. The malonate is used here in place of the oxalate of the oxaliplatin because an additional carbon atom is required to create a bond with the tamoxifen skeleton.
 | ||
| Scheme 1 Synthesis of (DACH)Pt–tamoxifen derivatives. | ||
 | ||
| Chart 6 (DACH)Pt–tamoxifen derivatives. | ||
Feasibility studies to assess the binding of these complexes to ERα have been accomplished. We found for 6, which contains a phenol group, an RBA of 6.4%, while molecule 5, deprived of this phenolic OH, showed a decreased recognition (RBA = 0.5%). A similar discrepancy was observed between OH-TAM (RBA = 37%) and tamoxifen (RBA = 1%).
The proliferative/antiproliferative effects of compounds 5, 6 and (DACH)PtCl2, 7, on the MCF7 breast cancer cell line were evaluated. The values given in Fig. 1 are expressed as a percentage of DNA present after 5 day incubation, compared with the control value of 100%. Complex 6 shows a clear antiproliferative activity on the MCF7 cells, with an IC50 value of 4.0 μM. This value is better than our results for the unvectorised (DACH)PtCl2 compound (IC50 = 6.3 μM), as well as that reported in the literature for oxaliplatin (IC50 = 7.4 μM), while the effect found for 5 is weaker (IC50 = 14 μM). Insofar as compound 5, deprived of the hydroxyl group, is less well recognised by the receptor, it appears that the effect observed with these complexes is primarily antioestrogenic, while the antiproliferative effect observed with (DACH)PtCl2 can be attributed to the recognised cytotoxicity of the metal group. At these concentrations, on the order of 10−6 M, platinum does not bring any particular benefit, and thus these molecules cannot be defined as new beneficial SERMs for the treatment of breast cancer. It is thus advisable to turn to the context of other metal systems.
 | ||
| Fig. 1 Antiproliferative effects of OH-TAM (1 × 10−7 M), 5, 6 and 7 (DACH)PtCl2 on MCF7 cells after 5 days of culture. The values in parentheses correspond to the log of the molarity of incubation. | ||
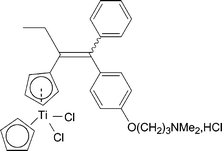 | ||
| Chart 7 Titanocifen. | ||
The synthesis of molecule 12, requiring the fixing of titanium in the last step of the process, was very difficult. Therefore, a new preparative process was developed, clarified in Scheme 2.51
 | ||
| Scheme 2 Synthetic strategy for production of titanocifens. | ||
The propionyl cyclopentadienyl manganese tricarbonyl 8 and ketone 9 were combined in the presence of a McMurry reagent to give the cross coupling product 10 with a yield of 54% and a 50/50 diastereomeric ratio. The innovative step in this synthesis is the photochemical decomplexation of the manganese moiety to form the cyclopentadienyl 11 with a 98% yield. The generation of the cyclopentadiene 11 was accomplished by the action of the protic solvent via photochemical exchange of CO for MeOH followed by intramolecular proton transfer according to Scheme 3. The CpTiCl3 moiety was then complexed to 11 with 57% yield. This new decomplexation reaction of cyclopentadienyl compounds gives access to polyfunctional organometallic cyclopentadienes, and has a real synthetic value because of the high yields and the mildness of the reaction conditions.
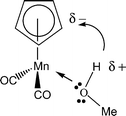 | ||
| Scheme 3 | ||
The titanium complex 12 recognises ERα rather well, in spite of the absence of a hydroxyl group, with an RBA value of 8.5%. The lipophilicity value of 12 was found to be 5.83 for the Z isomer and 5.95 for the E isomer. These values are quite high; estradiol presented a logPo/w of only 3.5.
The effect of 12 on hormone dependent MCF7 cells, observed after 6 days of culture, is shown in Fig. 2. The results are compared with those found in the presence of estradiol, which is the standard for the oestrogenic effect, and hydroxytamoxifen, the standard for the antioestrogenic effect. While E2 and OH-TAM yield their expected respective effects, 12 acts like an oestrogen almost as powerful as estradiol itself, even at very low concentrations (1 × 10−8 M). This intense oestrogenic effect was strong enough to conceal any antioestrogenic effect produced by the side chain carrying the dimethyl amino group. This unexpected behaviour led to the examination of the effect of the Cp2TiCl2 entity, 13, alone. In this case, we observed a proliferative behaviour even higher than that of 12. Therefore, it appears that the proliferative effect shown by compound 12 is due to the Cp2TiCl2 entity or one of its hydrolysis products.
 | ||
| Fig. 2 Effect of E2 (1 × 10−10 M), OH-TAM (1 × 10−7), 12, and 13 on MCF7 cells after 6 days of culture. The values in parentheses correspond to the log of the molarity of incubation. | ||
The hydrolysis of Cp2TiCl2 has been the subject of several studies52 and appears to proceed by a complex series of events which depend largely on conditions. A study by Sadler and co-workers48 suggests that the following process takes place in the biological environment. Initially the molecule undergoes hydrolysis of the Cl groups, which are replaced by H2O, yielding both mono- and dinuclear compounds. The hydrolysis of the cyclopentadienyl ligand, leading to cyclopentadiene, is a slower reaction which delays the complete release of Ti(IV). The metal can then bind transferrin, which may act as a vehicle for entry into the cell. It is thus necessary to consider the coordination chemistry of the Ti(IV) ion within the framework of possible coordination with the oestrogen receptor. The effects of some metal ions on ER mediated transcription have been reported in the literature. For example, the treatment of the receptor by Ni2+ or Cu2+ inhibited the association of the DNA binding domain of the receptor with the ERE.53 Additionally, the treatment of the receptor with very low concentrations of Cd2+ resulted in the coordination of this metal to the ER ligand binding domain and the demonstration of an oestrogenic effect.54 Several amino acid residues present in the ligand binding domain such as cysteines 381, 447, glutamic acid 523, histidine 524, and aspartic acid 538 are suitable for such coordination. A molecular modelling study (Fig. 3) with Ti4+ showed that cysteine 381 and glutamic acid 380 of helix 4 and histidine 547 of helix 12 are well positioned to close the gap between helices 4 and 12 and activate the receptor, in the same manner that occurs in the presence of estradiol. Although this mechanism is only hypothetical as yet, the reality of the oestrogenic effect caused by the titanocene compounds is indisputable. As such, our initial objective was not achieved, and it appears problematic to use complexes of Ti4+ in the fight against breast cancer. It should also be noted that these results raise questions about the role of titanium salts in environmental problems.
 | ||
| Fig. 3 Results of molecular modeling study with Ti4+. | ||
The molecule 14a depicted in Chart 8, first synthesised in 1996, was the first organometallic-tamoxifen SERM that, to our knowledge, presented promising biochemical data in terms of receptor recognition and aqueous stability.56 However, in vivo, on atthymic nude mice xenografted with T47D breast cancer cells, this molecule caused a large increase in the tumour volume (50% in fifteen days). With the idea that 14a was not deforming the ER to the “open” conformation, this problem was solved by increasing the length of the amino chain by one or two carbon atoms.57
 | ||
| Chart 8 Hydroxyferrocifens 14a–e. | ||
Again, we found that the most efficient path to the hydroxyferrocifens has as a key step a McMurry cross coupling reaction starting from the ferrocenyl ketone, outlined in Scheme 4. In this way, a series of ferrocifens and hydroxyferrocifens with n = 2, 3, 4, 5 and 8 were easily obtained as mixtures of Z and E geometrical isomers. These products were separated in certain cases and characterised by NMR and X-ray crystallography. The interconversion between the Z and E isomers is particularly rapid in protic solvents, but very slow, even nonexistent, in solvents with high pKa values, such as DMSO (pKa = 31) and benzene (pKa = 37).58 We interpreted this result by positing the formation of an intermediate α-carbenium ion which was stabilised by electron donation from the ferrocenyl group. Because protic solvents would be expected to reproduce biological (aqueous) conditions better than non-protic solvents, the majority of the biological tests were carried out with a mixture of Z and E isomers, although, in certain cases, we verified that the Z isomer was recognised by the receptor better than the E isomer. This type of E/Z isomerisation also exists in the tamoxifen series, although isomerisation occurs at a much slower rate.
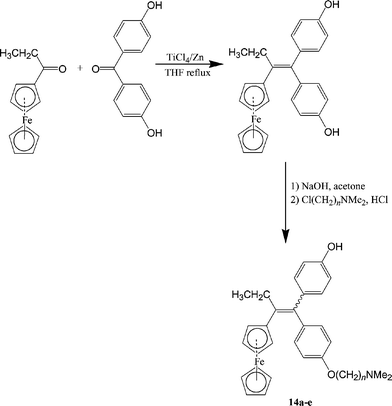 | ||
| Scheme 4 Synthesis of hydroxyferrocifens. | ||
The RBA values of various hydroxyferrocifens are presented in Table 1. The values obtained were lower for the hydroxyferrocifens than for hydroxytamoxifen, undoubtedly due to the steric effect of the ferrocenyl group compared to that of the phenyl group in OH-TAM. These values also decreased with the lengthening of the basic chain. Nonetheless, for the number of carbons ranging between n = 2 and n = 5, recognition was completely satisfactory. Measurements of octanol/water partition coefficients consistently showed a higher lipophilicity for the hydroxyferrocifens versus that of OH-TAM due to the lipophilic ferrocene moiety and the increasing length of the carbonaceous chain.
Po/w values for hydroxyferrocifensa
| RBA (%) | |||||
|---|---|---|---|---|---|
| ERα | ERβ | ||||
| Compound | n | (0 °C, 3 h 30) | (25 °C, 3 h 30) | (0 °C, 3 h 30) | logPo/w |
| a All the tested compounds are mixture of both isomers (Z + E). Measurements were performed with stock solutions in DMSO. b Value by definition. c Mean of two experiments. d Mean of three experiments. | |||||
| Estradiol | 100b | 100b | 100b | 3.5 | |
| OH-TAM | 38.5 | n.d. | 24 | Z = 3.2 | |
| E = 3.4 | |||||
| 14a | 2 | 14.6 | n.d. | 10c | Z = 3.8 |
| E = 3.9 | |||||
| 14b | 3 | 11.5d | 9.2d | 12d | Z = 4.3 |
| E = 4.5 | |||||
| 14d | 5 | 5c | 4.6c | 6d | Z = 4.7 |
| E = 4.8 | |||||
| 14e | 8 | 2.3c | 2.4 | n.d. | Z = 6.0 |
| E = 6.3 | |||||
Results obtained on the antiproliferative effects of the hydroxyferrocifens, shown in Fig. 4, particularly with the products for which n = 3 and 5, presented the most marked effects and can be summarised in the following way. With MCF7 cells, the effect of the hydroxyferrocifens was very similar to that of hydroxytamoxifen, with the hydroxyferrocifens slightly more potent at a concentration of 0.1 μM, and superior at 1 μM. However, for the MDA-MB231 cells, classified as hormone independent, the effects are spectacularly different. The organometallic hydroxyferrocifens displayed remarkable antiproliferative behaviour while hydroxytamoxifen was completely inactive. In the case of hydroxyferrocifens, one can thus postulate two kinds of behaviour; one which resembles the antagonistic role of hydroxytamoxifen on the ER, and a second which involves the in situ activation of the organometallic function.
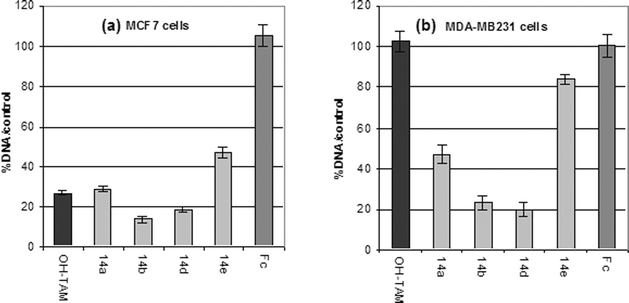 | ||
| Fig. 4 Effect of 1 μM OH-TAM, ferrocifens 14a, b, d and e, and ferrocene, Fc, after 5 days of culture on (a) MCF7 and (b) MDA-MB231 cells. | ||
Fig. 5, based on the structural study of the LBD of ER, described by Shiau et al., and using program MOLVIEW conceived by Cense,59 shows OH-ferrocifen (n = 4) residing in the binding site. It can be seen that the basic chain modifies the position of helix 12 of the receptor and provokes a similar antagonistic conformation to that of OH-TAM. Furthermore, there is the possibility of a stabilizing interaction between the chain nitrogen and the Asp351 residue. The use of the software Mac Spartan Pro makes it possible to obtain addition molecular modelling data,60 shown in Fig. 6. In this study, only the amino acids constituting the walls of the cavity were preserved. The hydroxyferrocifen 14b (n = 3) was digitally introduced into the cavity and the ideal position of the ligand was determined. By molecular mechanics using the Merck Molecular Force Field the variation of E of the complexation reaction of the bioligand with the cavity was calculated. For molecule 14b, where n = 3, one finds E = −52 kcal mol−1, while for n = 2, E is only equal to −41 kcal mol−1.
 | ||
| Fig. 5 Interactions of 14c docked in the ERα LBD. | ||
 | ||
| Fig. 6 Molecular modelling study of (a) 14b (Z) and (b) 14b (E). | ||
It is necessary to mention the fact that MCF7 cells contain primarily the α form of the oestrogen receptor, and to a much smaller degree, the recently discovered β receptor. While MDA-MB231 cells (previously classified as ER−) do not contain ERα, ERβ is, in fact, present, and has been found to be present in tamoxifen-resistant breast tumour cells as well. The role of the shape of the β receptor is the subject of intensive research.61 In particular, it has been theorised that molecules approximating a spherical structure are more readily recognised than planar molecules,62 which suggests that ERβ could be a target for the ferrocifens. Secondly, among the functions found for ERβ, one showing its role in the control of intracellular redox processes demands our attention.63 Ferrocene is easily oxidisable to the ferrocenium Fc+ radical cation, which can catalytically form superoxide ion O2− and the hydroxyl radical in the presence of water and O2.64 These oxidants are known to influence the apoptosis of cells and the generation of lesions on DNA. We do not currently have any direct evidence that ERβ is involved in the mechanism of action of the ferrocifens, but this possibility cannot be presently excluded.
Certain authors have observed that high concentrations of tamoxifen are cytotoxic to cancer cells,65 likely due to electrophilic species produced in its oxidative metabolism. It is possible that the increased sensitivity of hydroxyferrocifens to oxidation activates and enhances the process in concentrations of about 1 μM. In fact, electrochemical results that we have recently obtained suggest that the ferrocenium ion acts as an intramolecular oxidizing agent in basic conditions. The presence of an electron sink (ferrocenium) and the conjugated π-system combine to facilitate the proton-coupled oxidation of the phenol group, possibly generating an electrophilic quinone methide-type structure.66 Whatever the mechanism, these biochemical results place the ferrocifens in an area of concentration compatible with a potential therapeutic application and confer upon them a new character by combining a hormonal antagonist effect with a cytotoxic functionality.
The synthesis of the “ruthenocifen” series is shown in Scheme 5. The McMurry coupling reaction furnished mixtures of Z and E isomers of the bromo derivatives 15a–d, which were then converted to the amino compounds 16a–d. Unlike the ferrocifens, the ruthenocifen isomers interconverted too rapidly to be separated by HPLC. We suggested earlier that the intermediate species in the isomerisation mechanism is the α-carbenium ion formed by protonation of the double bond; therefore greater stability of this species would enhance isomerisation.70 This hypothesis is supported by the observation that the pKR+ value of [CpRu(η5-C5H4CH2)]+ is two orders of magnitude higher than that of the corresponding iron cation.71 This stability is likely conferred by stronger back bonding via the more diffuse Ru orbitals.72
 | ||
| Scheme 5 Synthesis of metallocifens where M = CpRu and Re(CO)3. | ||
The RBA values obtained on the α form of the receptor were remarkably high for the complexes with the side chain with n = 2 and 3, 85 and 53%, respectively. These values are higher than the RBA value found for OH-TAM (39%), which is unprecedented in our studies of other organometallic SERMs. The RBA values obtained for ERβ were also high, and in this case comparable to that found for OH-TAM. The ruthenocifens were also found to be more lipophilic than estradiol, increasing slightly with the length of the side chain.
On the MCF7 breast cancer cell line the hydroxyruthenocifen complexes showed an antiproliferative effect, Fig. 7(a). The effect of 16a was similar to that observed with OH-TAM, yet weaker than that of the three other ruthenium compounds 16b–d. However, to our disappointment, these compounds, like E2 and OH-TAM, had no effect on the MDA-MB231 breast cancer cell line. Therefore, the ruthenocene complexes of tamoxifen seem to act solely in an antioestrogenic fashion similar to OH-TAM, the organometallic moiety imparting no particular advantage. Electrochemical experiments showed that the oxidised compounds are highly unstable, indicating that these compounds do not likely form the putative reactive quinone methide, but decompose quickly in solution. The decomposition pathway has not yet been determined, but it is clear that the reactivity of these compounds is quite different than that of the ferrocifens.
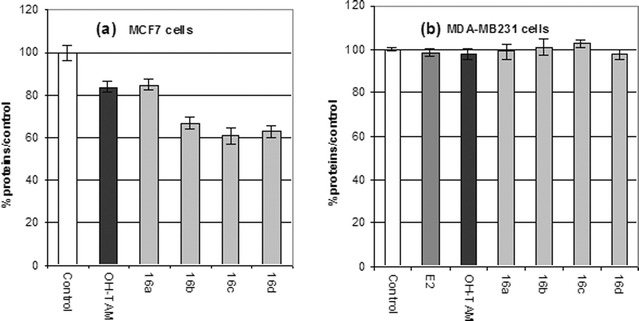 | ||
| Fig. 7 Antiproliferative effects of 1 μM of OH-Tam and of the ruthenocifens 16a–d on (a) MCF7 cells after 5 days of culture and (b) on MDA-MB231 cells after 6 days of culture. | ||
A series of CpRe(CO)3 tamoxifen derivatives have been synthesised via the McMurry coupling reaction with n = 2–5, 17a–d, Scheme 5. Contrary to the ferrocene and ruthenocene series, the separable Z and E rhenocifen isomers did not undergo interconversion in protic media. Insofar as the complex with n = 3 and 5 represented the best compromise between receptor affinity and antiproliferative effects, the following discussion will be limited to these molecules.
The RBA values, shown in Table 2, were quite satisfactory, and the lipophilicity values again surpassed that of hydroxytamoxifen. The results of study of the proliferative/antiproliferative effects of 17b and 17d on MCF7 and MDA-MB231 breast cancer cells are shown in Fig. 8. The rhenocifens were only slightly superior to OH-TAM, with the Z isomer modestly more active than the E isomer. With the MDA-MB231 cells no antiproliferative effect was observed for either isomer of either molecule.74
Po/w) of the rhenium derivatives of OH-TAM, (Z
+
E) mixtures, and separated Z and E isomersa
| ERα | ERα | ERβ | |||
|---|---|---|---|---|---|
| (cytosol) | (purified) | (purified) | log Po/wb | ||
| a Measurements performed with stock solutions in DMSO for 3 h at 0 °C. Mean of two experiments (general case) or three (when specified). b Mean of three experiments. | |||||
| 17a (n = 2) | (Z + E) | 6.4 ± 0.6 | 16.5 ± 1.1b | 9 ± 0.1b | |
| (Z) | 11.3 ± 0.9 | 28 ± 2 | 12 ± 3 | 4.5 | |
| (E) | 3.5 ± 0.1 | 4.0 ± 0.5 | 5.1 ± 0.8 | 4.7 | |
| 17b (n = 3) | (Z) | 12.3 ± 0.2 | 31 ± 3 | 16.8 ± 0.2 | 4.3 |
| (E) | 10.6 ± 0.7 | 11.2 ± 0.6 | 10.4 ± 0.4 | 4.5 | |
| 17c (n = 4) | (Z + E) | 8.5 ± 0.7b | 8.5 ± 0.5 | 6 ± 1 | |
| (Z) | 7.4 ± 0.2b | 22.6 ± 0.4 | 18 ± 1 | 4.4 | |
| (E) | 5.2 ± 0.6 | 3.6 ± 0.6 | 5.6 ± 0.1 | 4.6 | |
| 17d (n = 5) | (Z + E) | 2 ± 0.2 | — | — | |
| (Z) | 1.14 ± 0.05 | 16.0 ± 2.6b | 12 ± 0.2 | 4.7 | |
| (E) | 1.13 ± 0.05 | 7.2 ± 0.1 | 6.7 ± 0.1 | 5.0 | |
| 17e (n = 8) | (Z + E) | 1.0 ± 0.2 | — | 1.6 ± 0.1 | |
| (Z) | 1.3 ± 0.1 | 2.9 ± 0.5 | 5 ± 0.5 | 6.0 | |
| (E) | 0.7 ± 0.1 | 0.75 ± 0.05 | 2.7 ± 0.5 | 6.3 | |
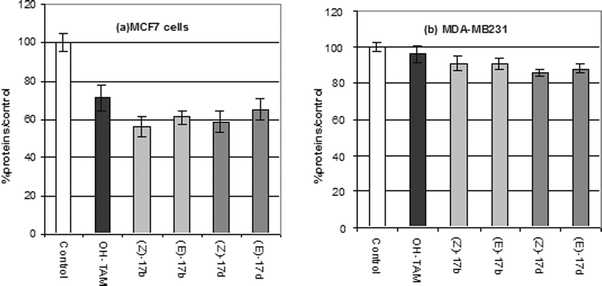 | ||
| Fig. 8 Antiproliferative effect of 1 μM of OH-TAM, (Z)-17b, (E)-17b, (Z)17d, (E)-17d on (a) MCF7 and (b) MDA-MB231 cells after 6 days of culture. | ||
A detailed molecular modelling study revealed the difference between the Z and E geometry inside the cavity. ER recognition was significantly better for the Z than for the E isomer, despite the fact that these two compounds have identical volumes, at 483 Å3 for 17b (Z) and 484 Å3 for 17b (E). Not surprisingly, the difference in affinity lies in the fact that the Z isomer is a better fit for the shape of the cavity than the E isomer. This molecular modelling study showed that 17b (Z) is associated with residues Arg394 and Glu353, while the position of Asp351 allowed the possibility of a hydrogen bond with the nitrogen of the basic chain O(CH2)3NMe2. The CpRe(CO)3 moiety which replaces the phenyl of OH-TAM was easily inserted in the area of His524 causing no notable perturbation. Fig. 9 shows the steric influence of the basic chain at the level of helix 12 of the protein, which can no longer bind to helix 4. This new position of H12, stabilised by the bond between Asp351 and the nitrogen of the NMe2 group of the side chain, is responsible as with OH-TAM for the antagonist effect observed with the MCF7 cell line.
 | ||
| Fig. 9 Molecular modelling of (a) 17b (Z) and (b) 17b (E). | ||
Although no unusual antiproliferative effects were noted for either the ruthenium or rhenium compounds, the fact that they behave similarly to OH-TAM suggests that they can be functionalised for radio imaging or therapy with no adverse effects. An important objective is the preparation of a complex of the Cp99mTc(CO)3 or Cp188Re(CO)3 type. In particular the use of Alberto's reagents75 [(H2O)3Re(CO)3]+, [(H2O)3Tc(CO)3]+, or the novel exchange reaction between [Re(CO)6)]+ and the ferrocene derivatives76 are good candidates to reach this objective.
Conclusion
The results obtained with the SERMs of metal complexes were quite varied. In the case of (DACH)Pt tamoxifen, the antiproliferative effect obtained is primarily the result of the behaviour of the antagonistic vector, while the cytotoxic moiety showed no appreciable effects. On the other hand, the effect of a metal ion released in situ can be strongly oestrogenic, so much so that it masks the behaviour of the vector, as seen with the organometallic Ti compounds. There is also the situation where the stable organometallic entity is only a simple spectator, as with CpRe(CO)3, or where the compound is very unstable, viz. Cp2Ru, and the result in both cases is close to that of OH-TAM. There is finally the case where the organometallic entity behaves as a genuine cytotoxic agent which is added to the antagonistic properties of the vector. There is in this type of molecule an enhancement of the SERM which may prove useful in the treatment of tamoxifen-resistant cancers. These products deserve thorough studies because of the prospects thus offered. Furthermore, a literature survey shows that this concept is not limited to the case of ferrocifens. A topical example is that of the ferroquine, the ferrocenic equivalent of chloroquine, which effective against malaria on stocks resistant to chloroquine.77 No doubt, the combination of organometallic compounds and organic vectors constitutes the emergence of a new therapeutic concept.References
- (a) A. A. Yuzpe, J. Reprod. Med., 2002, 47, 967–973 Search PubMed; (b) F. E. Likis, J. Midwifery Women's Health, 2002, 47, 139–156 Search PubMed.
- (a) The Writing Group for the PEPI, J. Am. Med. Assoc., 1996, 276, 1389–1396 Search PubMed; (b) T. E. R. Brown, J. Pharm. Practice, 2003, 16, 164–169 Search PubMed; (c) I. R. Reid, R. Eastell, I. Fogelman, J. D. Adachi, A. Rosen, C. Netelenbos, N. B. Watts, E. Seeman, A. V. Ciaccia and M. W. Draper, Arch. Int. Med., 2004, 164, 871–879 Search PubMed.
- P. E. Belchetz, N. Engl. J. Med., 1994, 330, 1062–1071 CrossRef CAS.
- Writing Group for the Women's Health Initiative Investigators, J. Am. Med. Assoc., 2002, 288, 321–333 Search PubMed.
- (a) R. D. Brinton, CNS Drugs, 2004, 18, 405–422 Search PubMed; (b) W. J. Cutter, M. Craig, R. Norbury, D. M. Robertson, M. Whitehead and A. D. G. Murphy, N. Y. Acad. Sci., 2003, 1007, 79–88 Search PubMed.
- (a) E. D. Staren and S. Omer, Am. J. Surg., 2004, 188, 136–149 CrossRef; (b) R. Nandur, K. Kumar and A. C. Villablanca, Prev. Cardiol., 2004, 7, 73–79 Search PubMed.
- K. D. Miller and G. W. Sledge, Jr., Invest. New Drugs, 1999, 17, 417–427 CrossRef CAS.
- (a) V. C. Jordan, Breast Cancer Res. Treat., 1988, 11, 197–209 CrossRef CAS; (b) K. Dhingra, Invest. New Drugs, 1999, 17, 285–311 CrossRef CAS; (c) M. J. Meegan and D. G. Lloyd, Curr. Med. Chem., 2003, 10, 181–210 CAS.
- A. Jemal, L. X. Clegg, E. Ward, L. A. G. Ries, X. Wu, P. M. Jamison, P. A. Wingo, H. L. Howe, R. N. Anderson and B. K. Edwards, Cancer, 2004, 101, 3–27 CrossRef.
- (a) J. Zidan, Z. Keidar, W. Basher and O. Israel, Med. Oncol., 2004, 21, 117–122 Search PubMed; (b) S. Turken, E. Siris, D. Seldiri, E. Flaster, G. Hyman and R. Lindsay, J. Natl. Cancer Inst., 1989, 81, 1086–1088 CrossRef CAS; (c) R. R. Love, R. B. Mazess, H. S. Barden, S. Epstein, P. A. Newcomb, V. C. Jordan, P. P. Carbone and D. L. DeMets, N. Engl. J. Med., 1992, 326, 852–856 CAS.
- W. J. Gradishar and V. C. Jordan, Hematol. Oncol. Clin. N. Am., 1999, 13, 435–455 Search PubMed.
- H. B. Muss, Breast Cancer Res. Treat., 1992, 21, 15–26 CrossRef CAS.
- (a) S. Mosselman, J. Polman and R. Dijkema, FEBS Lett., 1996, 392, 49–53 CrossRef CAS; (b) G. B. Tremblay, A. Tremblay, N. G. Copeland, D. J. Gilbert, N. A. Jenkins, F. Labrie and V. Giguere, Mol. Endocrinol., 1997, 11, 353–365 Search PubMed; (c) G. G. Kuiper, E. Enmark, M. Pelto-Huikko, S. Nilsson and J.-A. Gustafsson, Proc. Natl. Acad. Sci. USA, 1996, 93, 5925–5930 CrossRef CAS; (d) J.-A. Gustafsson, J. Endocrin., 1999, 163, 379–383 Search PubMed.
- (a) P. Walter, S. Green, G. Greene, A. Krust, J. M. Bornert, J. M. Jeltsch, A. Staub, E. Jensen, G. Scrace and M. Waterfield, et al., Proc. Natl. Acad. Sci. USA, 1985, 82, 7889–7893 CAS; (b) S. Green, P. Walter, V. Kumar, A. Krust, J. M. Bornert, P. Argos and P. Chambon, Nature, 1986, 320, 134–139 CAS; (c) G. L. Greene, P. Gilna, M. Waterfield, A. Baker, Y. Hort and J. Shine, Science, 1986, 320, 1150–1154 CrossRef.
- (a) V. Speirs, A. T. Parkes, M. J. Kerin, D. S. Walton, P. J. Carleton, J. N. Fox and S. L. Atkin, Cancer Res., 1999, 59, 525–528 CAS; (b) H. Dotzlaw, E. Leygue, P. H. Watson and L. C. Murphy, Cancer Res., 1999, 59, 529–532 CAS; (c) H. Sasano, T. Suzuki, Y. Matsuzaki, T. Fukaya, M. Endoh, H. Nagura and M. Kimura, J. Clin. Endocrinol. Metab., 1999, 84, 781–785 CrossRef CAS; (d) V. Speirs, C. Malone, D. S. Walton, M. J. Kerin and S. L. Atkin, Cancer Res., 1999, 59, 5421–5424 CAS; (e) S. A. Fuqua, R. Schiff, I. Parra, W. E. Friedrichs, J. L. Su, D. D. McKee, K. Slentz-Kesler, L. B. Moore, T. M. Willson and J. T. Moore, Cancer Res., 1999, 59, 5425–5428 CAS.
- (a) E. A. Vladusic, A. E. Hornby, F. K. Guerra-Vladusic and R. Lupu, Cancer Res., 1998, 58, 210–214 CAS; (b) Y. F. Hu, K. M. Lau, S. M. Ho and J. Russo, Int. J. Oncol., 1998, 12, 1225–1228 CAS.
- (a) E. Enmark, M. Pelto-Huikko, K. Grandien, S. Lagercrantz, J. Lagercrantz, G. Fried, M. Nordenskjold and J. A. Gustafsson, J. Clin. Endocrinol. Metab., 1997, 82, 4258–4265 CrossRef CAS; (b) M. Ponglikitmongkol, S. Green and P. Chambon, EMBO J., 1988, 7, 3385–3388 CAS; (c) L. Zhao and R. D. Brinton, J. Med. Chem., 2005, 48, 3463–3466 CrossRef CAS.
- (a) G. G. Kuiper, B. Carlsson, K. Grandien, E. Enmark, J. Haggblad, S. Nilsson and J.-A. Gustafsson, Endocrinology, 1997, 138, 863–870 CrossRef CAS; (b) T. Barkhem, B. Carlsson, Y. Nilsson, E. Enmark, J.-A. Gustafsson and S. Nilsson, Mol. Pharmacol., 1998, 54, 105–112 CAS; (c) M. J. Meyers, J. Sun, K. E. Carlson, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 1999, 42, 2456–2468 CrossRef CAS; (d) S. R. Stauffer, C. J. Coletta, R. Tedesco, G. Nishiguchi and K. Carlson, et al., J. Med. Chem., 2000, 43, 4934–4947 CrossRef CAS; (e) S. R. Stauffer, J. Sun, B. S. Katzenellenbogen and J. A. Katenellenbogen, Bioorg. Med. Chem., 2000, 8, 1293–1316 CrossRef CAS.
- (a) B. R. Henke, T. G. Consler, N. Go, R. L. Hale, D. R. Hohman, S. A. Jones, A. T. Lu, L. B. Moore, J. T. Moore, L. A. Orband-Miller, R. G. Robinett, J. Shearin, P. K. Spearing, E. L. Stewart, P. S. Turnbull, S. L. Weaver, S. P. Williams, G. B. Wisely and M. H. Lambert, J. Med. Chem., 2002, 45, 5492–5505 CrossRef CAS; (b) C. P. Miller, M. D. Collini and H. A. Harris, Bioorg. Med. Chem. Lett., 2003, 13, 2399–2403 CrossRef CAS.
- (a) R. E. Coleman, Cancer, 2003, 97(3 Suppl), 880–886 CrossRef; (b) M. Colleoni, A. Coates, O. Pagani and A. Goldhirsch, Cancer Treat. Rev., 1998, 24, 15–26 CrossRef CAS.
- E. J. Suh-Burgmann and A. Goodman, Annu. Int. Med., 1999, 131, 127–135 Search PubMed.
- Complete reviews of the mechanisms of ER regulated transcription are found in: (a) V. C. Jordan, J. Med. Chem., 2003, 46, 883–908 CrossRef; V. C. Jordan, J. Med. Chem., 2003, 46, 1081–1111 CrossRef; (b) K. C. Osborne, H. Zhao and S. A. W. Fuqua, J. Clin. Oncol., 2000, 18, 3172–3186.
- B. Hanstein, S. Djahansouzi, P. Dall, M. W. Beckmann and H. G. Bender, Eur. J. Endocrinol., 2004, 150, 243–255 Search PubMed , and references therein.
- (a) A. M. Brzozowski, A. C. Pike, Z. Dauter, R. E. Hubbard, T. Bonn, O. Engstrom, L. Ohman, G. L. Greene, J.-A. Gustafsson and M. Carlquist, Nature, 1997, 389, 753–758 CrossRef CAS; (b) A. K. Shiau, D. Barstad, P. M. Loria, L. Cheng, P. J. Kushner, D. A. Agard and G. L. Green, Cell, 1998, 95, 927–937 CAS; (c) A. C. W. Pike, A. M. Brzozowski, R. E. Hubbard, T. Bonn, A.-G. Thorsell, O. Engstrom, J. Ljunggren, J.-A. Gustafsson and M. Carlquist, EMBO J., 1999, 18, 4608–4618 CrossRef CAS.
- It is interesting to note that the DNA binding domain of the receptors contains zinc fingers (Zn2+), allowing it to complex with the ERE. However, a treatment of the receptor with another metal, for example Cu2+, prevents this association with the DN A. P. F. Predki and B. Sarkar, J. Biol. Chem., 1992, 267, 5842–5846 Search PubMed.
- K. Paech, P. Webb, G. G. Kuiper, S. Nilsson, J. Gustafsson, P. J. Kushner and T. S. Scanlan, Science, 1997, 277, 1508–1510 CrossRef CAS.
- (a) Y. Shang and M. Brown, Science, 2002, 295, 2465–2466 CrossRef CAS; (b) B. S. Katzenellenbogen and J. A. Katzenellenbogen, Science, 2002, 295, 2380–2381 CrossRef CAS.
- (a) Ref. 23; (b) P. Webb, G. N. Lopez, R. M. Uht and P. J. Kushner, Mol. Endocrinol., 1995, 9, 443–456 Search PubMed; (c) P. Webb, P. Nguyen, C. Valentine, G. N. Lopez, G. R. Kwok, E. McInerney, B. S. Katzenellenbogen, E. Enmark, J.-A. Gustafsson, S. Nilsson and J. Kushner, Mol. Endocrinol., 1999, 13, 1672–1685 Search PubMed.
- (a) G. Bèrubè, Y. He, S. Groleau, A. Sènè, H. M. Thèrieu and M. Caron, Inorg. Chim. Acta, 1997, 262, 139–145 CAS; (b) Y. He, S. Groleau, R. C. Gaudreault, M. Caron, H. M. Thèrien and G. Bèrubè, Bioorg. Med. Chem. Lett., 1995, 5, 2217–2222 CrossRef CAS.
- D. H. Hunter and L. G. Luyt, Bioconjugate Chem., 2000, 11, 175–181 CrossRef CAS.
- J. F. Valliant, P. Schaffer, K. A. Stephenson and J. F. Britten, J. Org. Chem., 2002, 67, 383–387 CrossRef CAS.
- A. Vessières, unpublished results.
- Y. Endo, T. Iijima, Y. Yamakoshi, H. Fukasawa, C. Miyaura, M. Inada, A. Kubo and A. Ital, Chem. Biol., 2001, 8, 341–355 CrossRef CAS.
- B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS.
- B. Lippert, Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, John Wiley and Sons, New York, 1999 Search PubMed.
- (a) E. von Angerer, in Metal Complexes in Cancer Chemotherapy, ed. B. K. Keppler, Wiley-VCH, Weinheim, Germany, 1993, pp. 73–83 Search PubMed; (b) J. Altman, T. Castrillo, W. Beck, G. Bernhardt and H. Schönenberger, Inorg. Chem., 1991, 30, 4085–4088 CrossRef CAS; (c) C. Chesne, G. Leclercq, P. Pointeau and H. Patin, Eur. J. Med. Chem., 1986, 21, 321–327 CAS; (d) R. Gust, H. Schńenberger, U. Klement and K. J. Range, Arch. Pharm. (Weinheim), 1993, 326, 967–976 CrossRef CAS; (e) J. Karl, R. Gust, T. Spruss, M. R. Schneider, H. Schńenberger, J. Engle, K. H. Wrobel, F. Lux and S. Trebert Haeberlin, J. Med. Chem., 1988, 31, 72–83 CrossRef; (f) O. Gandolfi, J. Blum and F. Mandelbaum-Shavit, Inorg. Chim. Acta, 1984, 91, 257–261 CrossRef CAS; (g) A. Jackson, J. Davis, R. J. Pither, A. Rodger and M. J. Hannon, Inorg. Chem., 2001, 40, 3964–3973 CrossRef CAS; (h) G. Grenier, B. Bèrubè and C. Gisquaud, Chem. Pharm. Bull., 1998, 46, 1480–1483 CAS.
- (a) H. Calvert, I. Judson and W. J. F. Van der Vijgh, Cancer Surv., 1993, 17, 189–217 Search PubMed; (b) M. C. Christian, Semin. Oncol., 1992, 19, 720–730 CAS; (c) L. R. Kelland, Crit. Rev. Oncol. Hematol., 1993, 15, 191–219 CrossRef CAS; (d) C. Meijer, H. Mulder, H. Timmer-Bosscha, W. J. Sluiter, G. J. Meersma and E. B. E. De Vries, Cancer Res., 1992, 52, 6885–6889 CAS.
- (a) V. C. Jordan, Tamoxifen for the Treatment and Prevention of Breast Cancer, PRR, New York, 1999 Search PubMed; (b) E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS.
- (a) P. J. O'Dwyer, J. Stevenson and S. W. Johnson, Drugs, 2000, 59(Suppl. 4), 19–27 CAS; (b) P. J. O'Dwyer, J. P. Stevenson and W. W. Johnson, in Cisplatin Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, Verlag Helvetica Chimica Acta, Zurich, Switzerland and Wiley-VCH, Weinheim, Germany, 1999, pp. 31–69 Search PubMed; (c) M. A. Jakupec, M. Galanski and B. K. Keppler, Rev. Physiol. Biochem. Pharmacol., 2003, 146, 1–53 Search PubMed.
- J. Reedijk, Chem. Commun., 1996, 801–806 RSC.
- V. Boudny, O. Vrana, F. Gaucheron, V. Kleinwachter, M. Leng and V. Brabec, Nucleic Acids Res., 1992, 20, 267–272 CrossRef CAS.
- (a) J. Lokich, Cancer Invest., 2001, 19, 756–760 CrossRef CAS; (b) J. Lokich and N. Anderson, Ann. Oncol., 1998, 9, 13–21 CrossRef CAS.
- (a) T. Andre, C. Boni, L. Mounedji-Boudiaf, M. Navarro, J. Tabernero, T. Hickish, C. Topham, M. Zaninelli, P. Clingan, J. Bridgewater, I. Tabah-Fisch and A. de Gramont, N. Engl. J. Med., 2004, 350, 2343–2351 CrossRef CAS; (b) S. G. Chaney, J. Oncol., 1995, 6, 1291–1305 Search PubMed; (c) Y. Kidani, K. Inagaki, M. Igo, A. Hashi and K. Kuretani, J. Med. Chem., 1978, 21, 1315–1318 CrossRef CAS; (d) G. Mathè, Y. Kidani, M. Segiguchi, M. Eriguchi, G. Fredj, G. Peytavin, J. L. Misset, S. Brienza, F. De Vassals, D. Chenu and C. Bourut, Biomed. Pharmacother., 1989, 43, 237–250 CrossRef CAS; (e) P. Souliè, E. Raymond, E. Cvitkovic and S. Brienza, Bull. Cancer, 1997, 84, 665–673 Search PubMed; (f) T. Tashiro, Y. Kawada and S. Y. Y. Kidani, Biomed. Pharmacother., 1989, 43, 251–260 CrossRef CAS.
- E. Raymond, C. Buquet-Fagot, S. Djelloul, J. Mester, E. Cvitkovic, P. Allain, C. Louvet and C. Gespach, Anti-Cancer Drugs, 1997, 8, 876–885 CrossRef CAS.
- S. Top, E. B. Kaloun, A. Vessières, G. Leclercq, I. Laïos, M. Ourevitch, C. Deuschel, M. J. McGlinchey and G. Jaouen, ChemBioChem, 2003, 4, 754–761 CrossRef CAS.
- J. E. McMurry, Acc. Chem. Res., 1983, 16, 405–411 CrossRef CAS.
- (a) M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511–2534 CrossRef CAS; (b) P. Köpf-Maier, Eur. J. Clin. Pharmacol., 1994, 47, 1 CAS; (c) J. B. Waern and M. M. Harding, J. Organomet. Chem., 2004, 689, 4655–4668 CrossRef CAS.
- M. Guo, H. Sun, H. J. McArdle, L. Gambling and P. J. Sadler, Biochemistry, 2000, 39, 10023–10033 CrossRef CAS.
- M. Guo, Z. Guo and P. J. Sadler, J. Biol. Inorg. Chem., 2001, 6, 698–707 Search PubMed.
- (a) K. Kröer, U. R. Kleeberg, K. Mross, L. Edler and D. K. Hossfeld, Onkologie, 2000, 23, 60–62 Search PubMed; (b) K. Mross, P. Robben-Bathe, L. Edler, J. Baumgart, W. E. Berdel, H. Fiebig and C. Unger, Onkologie, 2000, 23, 576–579 Search PubMed.
- S. Top, E. B. Kaloun, A. Vessières, I. Laïos, G. Leclercq and G. Jaouen, J. Organomet. Chem., 2002, 643–644, 350–356 CrossRef CAS.
- (a) H. D. Toney and T. Marks, J. Am. Chem. Soc., 1985, 107, 947–953 CrossRef CAS; (b) G. Mokdsi and M. M. Harding, J. Organomet. Chem., 1998, 565, 29–35 CrossRef CAS; (c) K. Doeppert, J. Organomet. Chem., 1987, 319, 351–354 CrossRef CAS.
- M. B. Martin, R. Reiter, T. Pham, Y. R. Avellanet, J. Camara, M. Lahm, E. Pentecost, K. Pratap, B. A. Gilmore, S. Divekar, R. S. Dagata, J. L. Bull and A. Stoica, Endocrinology, 2003, 144, 2425–2436 CrossRef CAS.
- (a) A. Stoica, B. S. Katzenellenbogen and M. B. Martin, Mol. Endocrinol., 2000, 14, 545–553 Search PubMed; (b) M. D. Johnson, N. Kenney, A. Stoica, L. Hilakivi-Clarke, B. Singh, G. Chepko, R. Clarke, P. F. Sholler, A. A. Lirio, C. Foss, R. Reiter, B. Trock, S. Paik and M. B. Martin, Nat. Med., 2003, 9, 1081–1084 CrossRef CAS.
- (a) A. M. Joy, D. M. L. Goodgame and J. I. Stratford, Int. J. Radiat. Oncol. Biol. Phys., 1989, 16, 1053–1056 CAS; (b) D. Osella, M. Ferrali, P. Zanello, F. Laschi, M. Fontani, C. Nervi and G. Cavigiolio, Inorg. Chim. Acta, 2000, 306, 42–48 CrossRef CAS; (c) H. Tamura and M. Miwa, M., Chem. Lett., 1997, 11, 1177–1178 CrossRef.
- S. Top, J. Tang, A. Vessieres, D. Carrez, C. Provot and G. Jaouen, Chem. Commun., 1996, 955–956 RSC.
- G. Jaouen, S. Top, A. Vessières, G. Leclercq, J. Quivy, L. Jin and A. Croisy, C. R. Acad. Sci. Paris, Ser. IIc, 2000, 3, 89–93 CrossRef CAS.
- S. Top, A. Vessieres, G. Leclercq, J. Quivy, J. Tang, J. Vaissermann, M. Huche and G. Jaouen, Chem. Eur. J., 2003, 9, 5223–5236 CrossRef CAS.
- J. M. Cense, Phys. Theor. Chem., 1990, 71, 763–766 Search PubMed.
- Mac Spartan Pro, Wavefunction Co., Irvine, CA 92612, USA Search PubMed.
- D. R. Compton, S. Sheng, K. E. Carlson, N. A. Rebacz, I. Y. Lee, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2004(47), 5872–5893 Search PubMed , and references therein.
- R. S. Muthyala, K. E. Carlson and J. A. Katzenellenbogen, Biorg. Med. Chem. Lett., 2003, 13, 4485–4488 CrossRef CAS.
- M. M. Montano, A. K. Jaiswal and B. S. Katzenellenbogen, J. Biol. Chem., 1998, 273, 25443–25449 CrossRef CAS.
- K. J. Nikula, J. D. Sun, E. B. Barr, W. E. Bechtold, P. J. Haley, J. M. Benson, A. F. Eidson, D. G. Burt, A. R. Dahl and R. F. Henderson, et al., Fundam. Appl. Toxicol., 1993, 21, 127–139 CrossRef CAS.
- (a) F. Zhang, P. W. Fan, X. Liu, L. Shen, R. B. Van Breemen and J. L. Bolton, Chem. Res. Toxicol., 2000, 13, 63–62 CrossRef; (b) D. A. Cameron, A. A. Ritchie, S. Langdon, T. J. Anderson and W. R. Miller, Breast Cancer Res. Treat., 1997, 45, 99–107 CrossRef CAS.
- E. A. Hillard, A. Vessieres, L. Thouin, G. Jaouen and C. Amatore, Angew. Chem., Int. Ed., 2005 Search PubMed , in press.
- (a) E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Curr. Top. Med. Chem., 2004, 4, 1525–1535 Search PubMed; (b) M. Galanski, V. B. Arion, M. A. Jakupec and B. K. Keppler, Curr. Pharm. Des., 2003, 9, 2078–2089 Search PubMed.
- (a) R. E. Morris, R. E. Aird, P. del S. Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS; (b) R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS; (c) C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396–1397 RSC.
- (a) A. J. Taylor and M. Wenzel, Xenobiotica, 1978, 8, 107–112 CrossRef CAS; (b) M. Wenzel, P. Asindraza and G. Schachschneider, J. Labelled Compd. Radiopharm., 1983, 20, 1061–1071 CrossRef CAS; (c) E. Stadlbauer, E. Nipper and M. Wenzel, J. Labelled Compd. Radiopharm., 1977, 13, 491–508 CrossRef CAS; (d) K. Hoffmann, B. Riesselmann and M. Wenzel, J. Labelled. Compd. Radiopharm., 1980, 17, 421–430 CrossRef CAS.
- P. Pigeon, S. Top, A. Vessières, M. Huché, E. A. Hillard, E. Salomon and G. Jaouen, J. Med. Chem., 2005, 48, 2814–2821 CrossRef CAS.
- J. Tang, S. Top, A. Vessières, N. Sellier, J. Vaissermann and G. Jaouen, Appl. Organomet. Chem., 1997, 11, 771–781 CrossRef CAS.
- S. Barlow, A. Cowley, J. C. Green, T. J. Brunker and T. Hascall, Organometallics, 2001, 20, 5351–5359 CrossRef CAS.
- G. Jaouen, S. Top, A. Vessières and R. Alberto, J. Organomet. Chem., 2000, 600, 23–36 CrossRef CAS.
- S. Top, A. Vessières, P. Pigeon, M.-N. Rager, M. Huchè, E. Solomon, C. Cabestaing, J. Vaissermann and G. Jaouen, ChemBioChem, 2004, 5, 1104–1113 CrossRef CAS.
- (a) R. Schibli, R. Alberto, J. Petrig, K. Ortner, H. Schmalle, B. Sigrist, J. Stahel, R. Scharzbach and P. A. Schubiger, J. Labelled Compd. Radiopharm., 2001, 44, S723–725; (b) J. Wald, R. Alberto, K. Ortner and L. Candreia, Angew. Chem., Int. Ed., 2001, 40, 3062–3066 CrossRef CAS.
- (a) S. Top, S. Masi and G. Jaouen, Eur. J. Inorg. Chem., 2002, 1848–1853 CrossRef CAS; (b) S. Masi, S. Top, L. Boubekeur, G. Jaouen, S. Mundwiler, B. Spingler and R. Alberto, Eur. J. Inorg. Chem., 2004, 2013–2017 CrossRef CAS.
- (a) C. Boit, Curr. Med. Chem. Anti-Infect. Agents, 2004, 3, 135–147 Search PubMed; (b) K. Chibale, J. R. Moss, M. Blackie, D. van Schalkwyk and P. J. Smith, Tetrahedron Lett., 2000, 41, 6231–6235 CrossRef; (c) P. Beadley, M. A. L. Blackie, K. Chibale, C. Clarkson, R. Meijboom, J. R. Moss, P. J. Smith and H. Su, Dalton Trans., 2003, 3046–3051 RSC.
| This journal is © The Royal Society of Chemistry 2006 |