Spectroscopy and dynamics of A [2B1] allyl radical†
Luca
Castiglioni
,
Andreas
Bach
and
Peter
Chen
*
Laboratory of Organic Chemistry, ETH Zurich, 8093 Zurich, Switzerland. E-mail: chen@org.chem.ethz.ch; Fax: +41 44 632 1280; Tel: +41 44 632 2898
First published on 5th May 2006
Abstract
The A [2B1] ← X [2A2] band system between 380 and 420 nm was observed in a supersonic jet expansion. The allyl radical was found to dissociate following electronic excitation, releasing a hydrogen atom. Monitoring the appearance of the hydrogen atom photoproduct as a function of the excitation laser wavelength, similar spectral features are observed as in earlier absorption experiments. Time- and frequency-resolved photoionization of the hydrogen atom product provides information on the unimolecular dissociation dynamics. The measured dissociation rates and kinetic energy releases of both allyl radical, C3H5, and partially deuterated allyl radical, C3DH4, suggest direct loss of the central hydrogen atom, leading to allene as the major product.
1. Introduction
Allyl radical, C3H5, is presently one of the best understood polyatomic radicals. It is the simplest π-conjugated hydrocarbon radical, thus serving as a model system for a whole class of molecules.Small hydrocarbon radicals play a key role in combustion chemistry, in hydrocarbon crackers and in certain regions of the atmosphere as well as in interstellar space. All hydrocarbons with a framework of three carbon atoms are assumed to be precursors in the formation of polycyclic aromatic hydrocarbons and soot.1 Allyl is also an important intermediate in propane-, butane- and acetylene-rich flames.2 Kinetic modelling of combustion processes is very sensitive to the rates of reactions that produce or consume hydrogen atoms.3 Thus, data on the unimolecular dissociation of allyl is important.
Spectroscopy and dynamics of allyl radical have been studied for many years. Numerous absorption measurements have been performed in the visible and ultraviolet region.4–13 Whereas the spectroscopy of the B [2A1], C [2B1] and D [2B2] electronically-excited states were studied in detail earlier in our group using resonance-enhanced multiphoton ionization (REMPI),8,9 relatively little is known about the first electronically-excited A [2B1] state.
Nearly 40 years ago, Ramsay and Currie observed a weak absorption in the spectral range between 370–410 nm, with a band origin at 408.3 nm, after flash photolysis of various allylic compounds in the gas phase. They assigned this to a transition between the ground state and first excited state of the allyl radical.11 This was recently confirmed in two cavity ring-down experiments,4 as well as in a picosecond time-resolved experiment.14 Due to the diffuse nature of the observed bands, they were not assigned further and the homogenous broadening was attributed to either predissociation11 or isomerization to cyclopropyl radical.15,16
Although the allyl radical is stable in the electronic ground state, it becomes reactive following electronic excitation. Photodissociation studies of the B, C and D excited states performed in our group suggest rapid internal conversion to hot ground state allyl radicals, which eventually dissociate into a hydrogen atom and C3H4 on the ground state surface. Three main reaction channels were identified, which lead to a hydrogen atom and either allene, cyclopropene or propyne as photoproducts.17,18 The lifetimes of these three excited states have been measured by time-resolved photoelectron spectroscopy (PES) and were found to be in the range of 10–20 ps.14,19 In the same experiment, the authors found a resonant enhancement of the pump–probe signal upon excitation in the range between 400–410 nm. The observed signal decay, however, was limited by the instrument response function, thus a lifetime <5 ps of the A-state may be presumed.
The allyl B, C, and D excited states have a partially rotationally-resolved spectrum, which allowed the investigation of angular-momentum-selected dynamics.20 An investigation of J-dependent dynamics was of interest since Szpunar et al. reported allyl radicals that were unusually stable towards dissociation.21,22 They used photofragment translational spectroscopy to disperse rotationally and vibrationally energized radicals and found a considerable fraction with internal energies up to 15 kcal mol−1 above the barrier but that did not dissociate. They attributed the increased stability of these radicals to a centrifugal barrier. In our earlier angular-momentum-selected experiment, we could not find any observable difference between the photodissociation dynamics of high- and low-J allyl radicals at 115 kcal mol−1 total energy.20 The A-state therefore offers a good opportunity to probe dissociation dynamics at energies closer to the barrier to unimolecular dissociation and in the same energy range as in the study of Szpunar et al.
2. Experimental
All experiments were carried out in a standard molecular beam apparatus, equipped with a time-of-flight mass spectrometer. The details were previously described in an earlier publication,17 and we will only give a brief summary. The signal-to-noise ratio was increased by modification of the ion source, ion optics and detector of the mass spectrometer and improved stability of the dye lasers.20We generated a clean pulsed beam of cold allyl radicals by supersonic jet flash pyrolysis23 of allyl iodide, obtained from Aldrich and used without further purification, seeded in 1.6 bar of helium. Isotopically-labeled precursors were synthesized according to procedures given in the literature.8 The skimmed radical beam was crossed in the ionization chamber by two counter-propagating laser beams. Fig. 1 depicts the energetics of the pump–probe experiments. The two-fold expanded output of a Nd:YAG-pumped dye laser (Radiant Dyes Narrow Scan) was used for excitation. The 365 nm output of another dye laser was frequency tripled in a Kr-cell (220 mbar) to provide 121.6 nm VUV light for the 1s–2p transition in the hydrogen atom. The 2P-hydrogen atoms were ionized by the residual 365 nm light in the source of the time-of-flight mass spectrometer and detected on double channel plates. Two digital delay generators (Stanford Research DG 535) controlled the relative timing of the lasers and the valve with a resolution better than 2 ns. The data were recorded on a digital storage oscilloscope, averaged over typically 200 laser shots, and transferred to a computer.
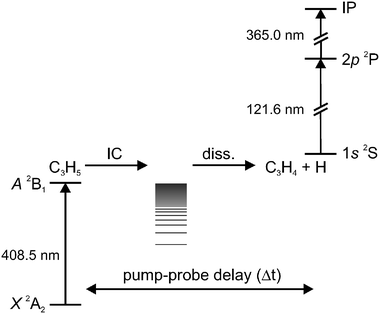 | ||
| Fig. 1 Schematic energy level diagram for the pump–probe experiments. After electronic excitation, the allyl radicals decay non-radiatively to form hot ground state radicals that dissociate into C3H4 and hydrogen. The hydrogen atoms are ionized by 1 + 1′ REMPI. | ||
We performed the following three types of experiment: (1) The probe laser was held fixed at the Lyman-α wavelength while the excitation laser was scanned in frequency to obtain an action spectrum of the hydrogen atoms; (2) the excitation laser was held fixed at a selected band of the A-state allyl while the probe laser was scanned in frequency to record the Doppler profiles of the hydrogen atoms; and (3) the wavelength of both lasers was fixed while the relative timing between them was changed to acquire a transient spectrum. The pump–probe delay in experiments (1) and (2) was set to 100 ns.
3. Ab initio calculations
Equilibrium geometries for both the ground and excited state were optimized using the complete active space self-consistent field (CASSCF) method24 with the cc-pVTZ, aug-cc-pVTZ, 6-311G(3df,3pd) and 6-311+G(3df,3pd) basis sets.25–27 A proper analysis of orbital occupancy showed that a small active space consisting of three electrons distributed among the three π-orbitals (1b1, 1a2, 2b1) is sufficient to account for the multireference character of the allyl radical. Harmonic frequency analysis in the first excited state was also performed at the CASSCF level. Energies were calculated using the multiconfiguration reference internally contracted configuration interaction (MRCI) method28,29 and extrapolated to the basis set limit (cc-pVXZ, X = D,T,Q).The geometries of stationary points on the C3H5 ground state surface and the possible dissociation products of allyl radical were optimized at the CCSD/cc-pVTZ level, whereas single-point energies were calculated including non-iterative triples excitation30,31 and extrapolated to the basis set limit (cc-pVXZ, X = D,T,Q). Vibrational frequencies used for zero-point energy correction and RRKM calculations were obtained using an anharmonic frequency analysis32 at the HCTH147/TZ2P level.33,34 All calculations were carried out using the MOLPRO35 and Gaussian 0336 packages.
4. Results and discussion
4.1 Vibrational structure of the A [2B1] ← X [2A2] band
Recording absorption spectra in a molecular beam was not feasible due to the inherently small number of radicals in the beam (∼1010 cm−3) and the low absorption cross section of the A [2B1] ← X [2A2] transition (∼2 × 10−19 cm2 molecule−1 at 402.9 nm).4 The oscillator strength associated with this transition (fosc = 0.0013) is more than two orders of magnitude lower than that observed for the UV bands (fosc = 0.26)7 and, combined with the presumed short lifetime of the A-state, also made resonance-enhanced multiphoton ionization (REMPI) spectroscopy impossible. Calculated reaction barriers for unimolecular reactions of the allyl radical suggest that the loss of hydrogen is possible at the energy of the A-state (∼70 kcal mol−1). We therefore monitored the appearance of hydrogen atoms as a function of excitation laser wavelength and obtained the action spectrum shown in Fig. 2. This spectrum shows the same spectral features as in the earlier absorption experiments by Currie and Ramsay11 and Tonokura and Koshi;4 a clear indication that these hydrogen atoms were released following excitation to the allyl A-state.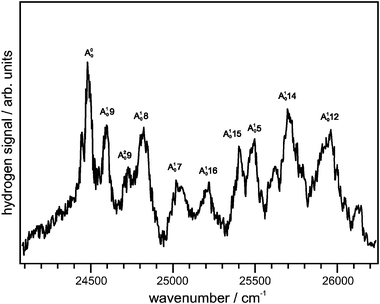 | ||
| Fig. 2 Action spectrum obtained by monitoring the total flux of hydrogen atoms while scanning the excitation laser. The delay time between excitation and probe laser was 100 ns. | ||
The excited states of the allyl radical have been the subject of numerous quantum chemical calculations.37–40 Noteworthy is an early LCAO self-consistent orbitals calculation by Longuet-Higgins and Pople.40 They calculated vertical excitation energies of 2.74 and 5.29 eV for the first two doublet excited valence states, and predicted a weak absorption for the transition in the visible region and a much stronger absorption in the UV. This has been experimentally confirmed and these two band systems correspond to transitions to the A- and C-excited states. We calculated a zero-point energy-corrected adiabatic excitation energy of the A [2B1] ← X [2A2] transition at the MRCI/CBS level of 3.04 eV, which is in quantitive agreement with the experiment. The configuration state function of the A-state can be described as a linear combination of the following two reference configurations: (1b1)1(1a2)2(2b1)0 and (1b1)2(1a2)0(2b1)1. This corresponds formally to either a π → n or a n → π* transition. It should be pointed out that the allyl C-state is a linear combination of the same reference configurations, but with different coefficients for each configuration.
The A [2B1] ← X [2A2] transition is allowed by electronic selection rules in both C2v and C2 symmetry. If a transition between states with different symmetry to the equilibrium conformation is considered, the selection rules must be applied to the common elements of symmetry, which is C2 in this case. The dipole transition moment lies along the long axis of the molecule (y) if the transition is treated in C2v symmetry, leading to an A-type band. Within C2 symmetry, the transition dipole moment lies in the xy-plane, leading to an A,C-type hybrid band. Fig. 3 shows a simulation of the rotational contour performed using the AsyrotWin program.41 The rotational constants for the allyl A-state equilibrium geometry were taken from ab initio calculations at the CASSCF/6-311+(3df,3pd) level of theory. The rotational temperature was taken from a rotational contour analysis of a REMPI-spectrum of the partially rotationally-resolved C-state obtained under the same experimental conditions, and was found to be ∼100 K. The simulation of an A,C-type hybrid band with a line FWHM of 2 cm−1 gave the best fit of the peak at 408.5 nm.42 The band origin is ∼10 cm−1 red-shifted from the peak maximum. The intervals between this origin and the vibronic bands observed in the action spectrum are listed in Table 1.
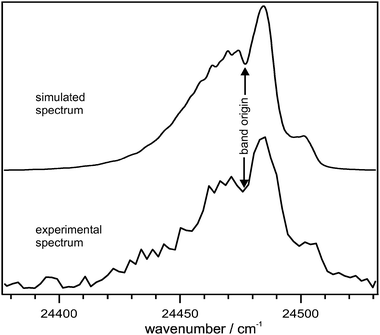 | ||
| Fig. 3 Simulation of the rotational contour in the A-state origin region as an A,C-type hybrid band. The rotational temperature was set to 100 K and a line FWHM of 2.0 cm−1 was used. | ||
| Mode | CASSCFa cc-pVTZ | CASSCFb 6-311+G (3df,3pd) | Expt.c | ||
|---|---|---|---|---|---|
| a Scaled by 0.910. b Scaled by 0.902. c Taken from the action spectrum depicted in Fig. 2, the uncertainty of the intervals is ≤ ± 10 cm−1. | |||||
| a | ν 1 | As CH2 str (in phase) | 3055 | 3029 | |
| ν 2 | CH str | 3003 | 2976 | ||
| ν 3 | Sym CH2 str (in phase) | 2966 | 2939 | ||
| ν 4 | Sym CH2 scis | 1439 | 1425 | ||
| ν 5 | Sym CH2 rock | 1095 | 1082 | 1032 | |
| ν 6 | Sym CCC str CH2 rock | 880 | 872 | (934) | |
| ν 7 | As CH2 wag | 511 | 509 | 553 | |
| ν 8 | CCC bend | 403 | 400 | 381 | |
| ν 9 | Sym CH2 twist | 160 | 156 | 135 | |
| b | ν 10 | As CH2 str | 3055 | 3028 | |
| ν 11 | Sym CH2 str | 2966 | 2939 | ||
| ν 12 | As CCC str | 1593 | 1572 | 1501 | |
| ν 13 | As CH2 scis | 1394 | 1382 | ||
| ν 14 | CH rock | 1230 | 1215 | 1232 | |
| ν 15 | As CH2 rock | 922 | 913 | 934 | |
| ν 16 | As CH2 twist CH wag | 750 | 751 | 752 | |
| ν 17 | Sym CH2 wag | 500 | 500 | ||
| ν 18 | CH wag (in phase) | 418 | 415 | ||
Harmonic frequencies were calculated at the CASSCF level for the C2 symmetric A-state equilibrium geometry. The frequency scaling factors were calculated by linear regression to experimental frequencies obtained from the tentatively assigned vibronic bands. The scaling factors are in the same range as derived from other calculations with these basis sets.43 The root-mean-square error of the scaled harmonic frequencies is 30.8 cm−1 for the 6-311+G(3df,3pd) basis set and 33.1 cm−1 for the cc-pVTZ basis set, which is much lower than the rms errors reported for various Hartree–Fock (HF) methods by Scott and Radom.43
Upon electronic excitation, the allyl radical undergoes significant changes in geometry. The C2 symmetric A-state equilibrium geometry has a non-planar structure, with the two terminal CH2 groups rotated out-of-plane by a dihedral angle of ∼40°. The CC bonds are substantially lengthened to 1.47 Å, while the remaining coordinates remain essentially unchanged. Both lengthening of the CC bond as well as the out-of-plane twisting of the CH2 groups can be explained by the promotion of electrons from the bonding π orbital to the non-bonding n or anti-bonding π* orbital. The ν9 asymmetric CH2 twist frequency is lowered from 547 cm−1 in the ground state to 135 cm−1 in the excited state. As this twist is one of the major changes in equilibrium geometry upon excitation, the ν9 mode is prominent in the spectrum and its first overtone also appears with about half of the intensity of the fundamental. Nearly all peaks observed in our action spectrum can be assigned to a vibrational mode of A-state allyl and tentative assignments are listed in Table 1. The nonplanar geometry results in a double-well potential for the ν9 mode. Tonokura and Koshi4 calculated the height of the inversion barrier to be around 400 cm−1, which is in agreement with our own calculations. This leads to an inversion doubling of levels at or below the top of the barrier, although the splittings are far too small to be observed in the experimental spectrum. The vibronic bands seem to broaden towards higher energy in the spectrum, possibly indicating a shortening of the lifetime.
4.2 Dissociation dynamics
As already discussed in the previous section, from the similarity between our observed action spectrum and earlier absorption spectra, as well as the agreement of experimental with calculated frequencies, it is evident that the observed hydrogen atoms originate from the A-state allyl. One question that may immediately arise is whether the dissociation occurs in the excited state or on the ground state surface. The photodissociation dynamics of the B, C, and D-states were examined in an earlier publication and it was found that dissociation occurred on the ground state surface after fast internal conversion.17,18 There are clear indications that the lifetime of the A-state is extremely short, presumably <5 ps, and we would thus not expect a dissociation in the excited state to proceed on a ns time scale as depicted in Fig. 4. We obtained the appearance time of hydrogen atoms by monitoring the total flux of hydrogen atoms while varying the time delay between excitation and probe laser. The data points were fitted using the expression| SH(t) = N(e−k1t − e−kHt) | (1) |
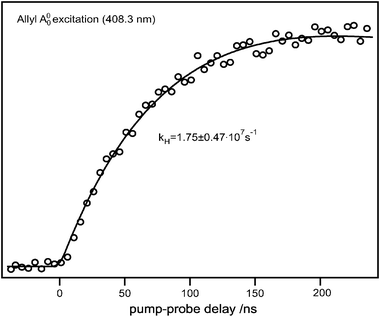 | ||
| Fig. 4 Appearance of the hydrogen atom signal as a function of the time delay between pump and probe laser for initial excitation to the A-state origin. | ||
Fig. 5 shows calculated reaction channels for allyl radical unimolecular dissociation. The lowest barrier is for cyclization to cyclopropyl radical via transition state 5. At 70 kcal mol−1 there is not enough energy available for allyl to dissociate to cyclopropene and a hydrogen atom. On the right hand side of the scheme, there is, in principle, enough energy available for any reaction to take place, leading to allene and propyne as final products. As TS 1 is more than 2 kcal mol−1 lower in energy than TS 2, we expect mainly allene and hydrogen as products.
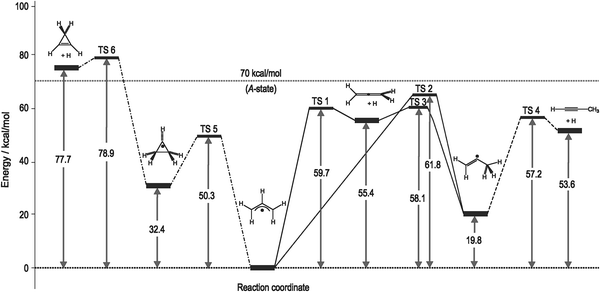 | ||
| Fig. 5 Possible reaction pathways for allyl radical unimolecular dissociation. Cyclization to cyclopropyl and subsequent dissociation to cyclopropene is depicted on the left-hand side. Direct loss of hydrogen to form allene via TS 1, or 1,2-H-shift leading to 2-propenyl, which can then dissociate to either allene or propyne, are shown on the right-hand side. Given energies are zero-point energy corrected and are calculated at CSSD(T)/cc-pVXZ (X = D,T,Q, extrapolated to basis set limit) level of theory at CSSD/cc-pVTZ geometries of these stationary points. | ||
The substitution of selected hydrogen atoms with deuterium allows an investigation of the site selectivity of the reaction. For the dissociation forming allene via TS 1, one would expect loss of the hydrogen connected to the central carbon atom in allyl. Upon cyclization to cyclopropyl on the other hand, one of the terminal C–H bonds should be cleaved. Finally, 2-propenyl formed by a 1,2-hydrogen shift can either dissociate to propyne or allene, giving rise to isotopic scrambling. In order to identify the dominant channel, 2-deuterioallyliodide (in which the central hydrogen atom is replaced by deuterium) was synthesized. We performed Doppler spectroscopy of the hydrogen and deuterium atom photofragments and obtained the spectrum depicted in Fig. 6. The 1s–2p Lyman-α transition in deuterium is blue-shifted only by 22.4 cm−1 with respect to hydrogen, so that both transitions could be recorded in the same scan of the probe laser. Due to the proximity of the signals, we assume the efficiency of the non-resonant frequency tripling for the generation of VUV photons to be constant over the whole range. If it is also presumed that both absorption and ionization cross sections for hydrogen and deuterium are comparable, the area under the H and D peaks is directly proportional to the number of H and D atoms lost from the parent allyl radical.
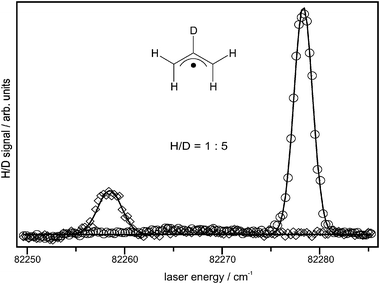 | ||
| Fig. 6 Doppler profiles for 2-deuterioallyl obtained from the A-state origin at a time delay of 100 ns between excitation and probe laser. The ratio between hydrogen (◇) and deuterium (○) is ∼1 ∶ 5. | ||
If the bond between deuterium and the central carbon is broken, one has to consider a first order kinetic isotope effect. Substitution of hydrogen with deuterium leads to a decrease of zero-point energy and an increase in the density of states, caused by a lowering of the C–D frequency compared to C–H. As a result, reaction rates involving cleavage of a C–H bond will, in general, decrease upon deuteration up to one order of magnitude. From the ratio between the H- and D-signals obtained from the Doppler profiles in Fig. 6, we conclude that direct dissociation to allene from loss of the central deuterium atom is the dominant reaction channel. Note that the kinetic isotope effect works in favor of cleavage of the terminal C–H bonds. We also measured Doppler profiles at different pump–probe delays but did not observe a significant change in the H/D ratio, except for a moderate increase of the deuterium signal at longer pump–probe delays. This can easily be explained by the faster movement of the hydrogen atoms out of the probe laser detection volume, which becomes significant at longer delay times. The isotopic purity of the radical precursor 2-deuterioallyliodide was determined by NMR and is >95%. Hence, a significant fraction of the observed hydrogen atoms is due to isotopic impurity.
RRKM calculations for direct allene formation yielded a microcanonical rate constant of 1.96 × 107 s−1, which is in good agreement with our experimental unimolecular dissociation rate kH(E) = 1.75 (± 0.47) × 107 s−1. We also measured the rates for the isotopically-labelled species and obtained kD(E) = 1.09 (± 0.43) × 107 s−1, which is roughly two times slower as a consequence of the kinetic isotope effect. More dissociation constants obtained after excitation of allyl to different energies are listed in Table 2 together with the calculated RRKM values. The measured dissociation constants are in good agreement with the calculated RRKM values and, as expected, the reaction becomes faster with increasing excitation energy.
| Energya/kcal mol−1 | k H(E)b | k D(E)b | k H, RRKM(E)c | k D, RRKM(E)c | |
|---|---|---|---|---|---|
| a Corresponding to excitation to indicated bands. b Obtained from time-dependent appearance of H/D atom signal as shown in Fig. 4. c RRKM calculations based on anharmonic frequencies at HCTH147/TZ2P level. | |||||
| 70.0 | A000 | 1.75 | 1.09 | 1.96 | 1.08 |
| 71.6 | A107 | 3.49 | 2.04 | 3.90 | 2.13 |
| 73.5 | A1014 | 6.58 | 3.89 | 7.78 | 4.24 |
From frequency-resolved photoionization of the hydrogen atom photoproduct, we obtained the Doppler profile shown in Fig. 7. The profile is broadened by the VUV laser linewidth of 0.5 cm−1 and a Gaussian FWHM of 2.67 cm−1 resulted after proper deconvolution. The translational temperature of the hydrogen atom in the laboratory coordinate frame can be calculated using the expression
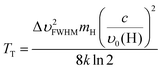 | (2) |
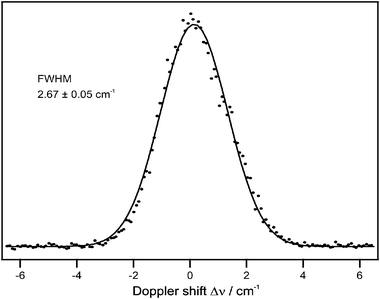 | ||
| Fig. 7 H-atom Doppler profile obtained following excitation into the A-state origin at 100 ns pump–probe delay. | ||
The high-energy wings of the Doppler profile in Fig. 7 may indicate a small contribution of higher-order multiphoton processes. We have measured the H-atom signal intensity at different excitation laser energies and found a linear dependence on laser power, typical for a one-photon process. Doppler profiles recorded at different laser intensities showed no significant difference. Furthermore, two-photon processes would lead to resonant Rydberg states around 200 nm with sharp bands, which were not observed in the spectrum.
4.3 2 A 2–2B1 conical intersections and the fate of the electronically-excited allyl radical
The short lifetime of the A-state may be explained in terms of the flat character of the potential energy surface along several internal coordinates, in particular the CH2-twist motion. An additional conrotatory 16° out-of-plane twisting of the terminal CH2 groups and a substantial decrease of the CCC angle leads to a conical intersection with the ground state surface, as shown in Fig. 8a, which lies only 1.10 kcal mol−1 higher in energy than the allyl A-state equilibrium geometry. Another conical intersection was located when the geometry was held in Cs symmetry and the CH2 groups were rotated in a disrotatory fashion (Fig. 8b). It should also be mentioned that both the C2 symmetric A-state equilibrium geometry and the minimum energy geometry at the conical intersection are chiral, and that there are two enantiomeric conformations with the terminal CH2 groups simply twisted the other way round. Dynamic correlation included at the MRCI/CBS level of theory at the CASSCF geometries leads to a ∼3 kcal mol−1 decrease in the minimum energy conical intersections relative to the A-state equilibrium geometry. These values should be considered with care, as one would expect different geometries of the minimum energy conical intersections at the MRCI level of theory. This is also reflected in the ∼0.5 kcal mol−1 energy differences of the upper and lower states, which are energetically exactly the same at the CASSCF level of theory.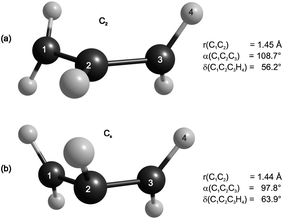 | ||
| Fig. 8 Geometries at the conical intersections between the A-state and ground state, located using SA-CASSCF/6-311+G(3df,3pd).44 The C2 geometry (a) is 1.1 kcal mol−1 higher and the Cs geometry (b) is 3.8 kcal mol−1 lower in energy than the A-state equilibrium geometry. | ||
An electrocyclic reaction between allyl and cyclopropyl radical has already been discussed when Woodward and Hoffmann introduced the concept of conservation of orbital symmetry.51 They suggested cyclization in a conrotatory fashion in the ground state and a disrotatory fashion in the excited state; however it was rapidly demonstrated by Longuet-Higgins that in both conrotatory and disrotatory modes the ground state of each radical is correlated to an excited state of the other using state correlation schemes,52 as depicted in Fig. 9. The issue of electrocyclic interconversion between allyl and cyclopropyl has been addressed by numerous theoretical studies. A quasi-classical trajectory study of the ring opening of cyclopropyl in the ground state via a highly asymmetric transition state resulted in nearly equal probability for conrotatory and disrotatory ring opening.53 A CASSCF study of the photochemical cyclization found no barrier on the conrotatory pathway and a very small (<1.5 kcal mol−1) barrier on the disrotatory pathway,16 which is in good agreement with the state correlation scheme in Fig. 9. Our own calculations show conical intersections along both pathways, and thus the excited allyl radical can either undergo cyclization leading to cyclopropyl or end up in the allyl ground state. Holtzhauer et al.15 found evidence for the formation of cyclopropyl after irradiation of allyl radicals in an argon matrix, which supports the picture of photochemical electrocyclization. The resulting cyclopropyl has enough internal energy to ring-open to allyl on the ground state surface again. This electrocyclic interconversion of allyl and cyclopropyl radical in the ground state is reversible and fast compared to the other unimolecular reactions shown in Fig. 5.
 | ||
| Fig. 9 State correlation scheme for conrotatory and disrotatory electrocyclic interconversion of cyclopropyl and allyl radical. Note that only valence excited states are considered in allyl, although there are some intruding low lying Rydberg states. | ||
5. Conclusions
We have measured the action spectrum of the allyl radical in the range of 380–420 nm. The observed vibronic bands correspond to those obtained in earlier absorption experiments and were assigned with the help of calculated vibrational frequencies for the A [2B1] electronically-excited state.The primary photophysical and photochemical processes upon excitation are very intricate. The three conical intersections of the A-state and the ground state and the possibility of electrocyclic conversion to cyclopropyl radicals give rise to various mechanisms for non-radiative decay, and thus explain the short lifetime of the A-state. Whereas we have no experimental information on the primary photochemical processes leading to hot ground state radicals, the measured kinetics of the hydrogen atom loss and Doppler profiles suggests that dissociation to the primary photoproducts, allene and a hydrogen atom, must occur on the ground state surface.
The observed diffuse spectrum unfortunately has no rotational resolution and prevents investigation of J-dependent dynamics at energies close to the barrier to unimolecular dissociation. A further investigation of the primary photochemical processes would demand sophisticated experiments, with isotopically-labeled precursors combined with extensive computational studies of the non-adiabatic dynamics.
Acknowledgements
We appreciate assistance in the early stage of the project and help in the synthesis of the deuterated precursor by Didier Zurwerra and Daniel Meier. The support of this work by the Schweizerischer Nationalfonds and ETH Zürich is gratefully acknowledged.References
- N. M. Marinov, M. J. Castaldi, C. F. Melius and W. Tsang, Combust. Sci. Technol., 1997, 128, 295 CrossRef CAS
.
- K. M. Leung and R. P. Lindstedt, Combust. Flame, 1995, 102, 129 CrossRef CAS
- J. A. Miller, R. J. Kee and C. K. Westbrook, Annu. Rev. Phys. Chem., 1990, 41, 345 CrossRef CAS
- K. Tonokura and M. Koshi, J. Phys. Chem. A, 2000, 104, 8456 CrossRef CAS
- E. Achkasova, M. Araki, A. Denisov and J. P. Maier, Mol. Phys., 2005, 103, 1555 CrossRef CAS
- A. D. Sappey and J. C. Weisshaar, J. Phys. Chem., 1987, 91, 3731 CrossRef CAS
- N. Nakashima and K. Yoshihara, Laser Chem., 1987, 7, 177 Search PubMed
- D. W. Minsek and P. Chen, J. Phys. Chem., 1993, 97, 13375 CrossRef CAS
- D. W. Minsek, J. A. Blush and P. Chen, J. Phys. Chem., 1992, 96, 2025 CrossRef CAS
- A. B. Callear and H. K. Lee, Trans. Faraday Soc., 1968, 64, 308 RSC
- C. L. Currie and D. A. Ramsay, J. Chem. Phys., 1966, 45, 488 CrossRef
- J. W. Hudgens and C. S. Dulcey, J. Phys. Chem., 1985, 89, 1505 CrossRef CAS
- J. A. Blush, D. W. Minsek and P. Chen, J. Phys. Chem., 1992, 96, 10150 CrossRef CAS
- T. Schultz, J. S. Clarke, H.-J. Deyerl, T. Gilbert and I. Fischer, Faraday Discuss., 2000, 115, 17 RSC
- K. Holtzhauer, C. Cometta-Morini and J. F. M. Oth, J. Phys. Org. Chem., 1990, 3, 219 CrossRef CAS
- M. Yamaguchi, J. Mol. Struct.: THEOCHEM, 1996, 365, 143 CrossRef CAS
- H.-J. Deyerl, I. Fischer and P. Chen, J. Chem. Phys., 1999, 110, 1450 CrossRef CAS
- H.-J. Deyerl, T. Gilbert, I. Fischer and P. Chen, J. Chem. Phys., 1997, 107, 3329 CrossRef CAS
- T. Schultz and I. Fischer, J. Chem. Phys., 1998, 109, 5812 CrossRef CAS
- L. Castiglioni, A. Bach and P. Chen, J. Phys. Chem. A, 2005, 109, 962 CrossRef CAS
- D. E. Szpunar, Y. Liu, M. J. McCullagh, L. J. Butler and J. Shu, J. Chem. Phys., 2003, 119, 5078 CrossRef CAS
- D. E. Szpunar, M. L. Morton, L. J. Butler and P. M. Regan, J. Phys. Chem. B, 2002, 106, 8086 CrossRef CAS
- D. W. Kohn, H. Clauberg and P. Chen, Rev. Sci. Instrum., 1992, 63, 4003 CrossRef CAS
- N. Yamamoto, T. Vreven, M. A. Robb, M. J. Frisch and H. B. Schlegel, Chem. Phys. Lett., 1996, 250, 373 CrossRef CAS
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS
- P. J. Knowles and H. J. Werner, Chem. Phys. Lett., 1988, 145, 514 CrossRef CAS
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1988, 89, 5803 CrossRef CAS
- J. D. Watts, J. Gauss and R. J. Bartlett, J. Chem. Phys., 1993, 98, 8718 CrossRef CAS
- J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968 CrossRef CAS
- V. Barone, J. Chem. Phys., 2004, 120, 3059 CrossRef CAS
- T. H. Dunning, J. Chem. Phys., 1971, 55, 716 CrossRef
- A. D. Boese, N. L. Doltsinis, N. C. Handy and M. Sprik, J. Chem. Phys., 2000, 112, 1670 CrossRef CAS
- H.-J. Werner, P. J. Knowles, R. Lindh, M. Schütz, P. Celani, T. Korona, F. R. Manby, G. Rauhut, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, C. Hampel, G. Hetzer, A. W. Lloyd, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, R. Pitzer, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni and T. Thorsteinsson, MOLPRO, a package of ab initio programs, Birmingham, UK, 2003.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, (Revision C.02), Gaussian, Inc., Wallingford CT, 2004 Search PubMed
- S. D. Peyerimhoff and R. J. Buenker, J. Chem. Phys., 1969, 51, 2528 CrossRef CAS
- F. Aquilante, K. P. Jensen and B. O. Roos, Chem. Phys. Lett., 2003, 380, 689 CrossRef CAS
- T.-K. Ha, H. Baumann and J. F. M. Oth, J. Chem. Phys., 1986, 85, 1438 CrossRef CAS
- H. C. Longuet-Higgins and J. A. Pople, Proc. Phys. Soc., London, Sect. A, 1955, 68, 591 CrossRef
- R. H. Judge and D. J. Clouthier, Comput. Phys. Commun., 2001, 135, 293 CrossRef CAS
- Achkasova et al. reported some rotational structure in a band they assigned to the A-state origin in their cavity ring-down experiment at a rotational temperature of ∼40 K (Ref. 5). J-Dependent lifetime broadening in the A-state (i.e. Coriolis coupling) at the higher rotational temperature in our experiment would lead to a broadened action spectrum arising from preferential dissociation of high-J radicals. Test simulations of the rotational band contour at our rotational temperature and omitting the low-J lines showed no significant change from the band depicted in Fig. 3.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502 CrossRef CAS
- F. Bernardi, M. A. Robb and M. Olivucci, Chem. Soc. Rev., 1996, 25, 321 RSC
- K. Tsukiyama and R. Bersohn, J. Chem. Phys., 1987, 86, 745 CrossRef CAS
- S. Satyapal, G. W. Johnston, R. Bersohn and I. Oref, J. Chem. Phys., 1990, 93, 6398 CrossRef CAS
- J. Park and R. Bersohn, J. Chem. Phys., 1990, 93, 5700 CrossRef CAS
-
R. D. Levine and R. B. Bernstein, Molecular Reaction Dynamics and Chemical Reactivity, Oxford University Press, New York, 1987 Search PubMed
- A. S. Sudbo, P. A. Schulz, E. R. Grant, Y. R. Shen and Y. T. Lee, J. Chem. Phys., 1979, 70, 912 CrossRef CAS
- D. H. Mordaunt, D. L. Osborn and D. M. Neumark, J. Chem. Phys., 1998, 108, 2448 CrossRef CAS
- R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395 CrossRef CAS
- H. C. Longuet-Higgins and E. W. Abrahamson, J. Am. Chem. Soc., 1965, 87, 2045 CrossRef CAS
- D. J. Mann and W. L. Hase, J. Am. Chem. Soc., 2002, 124, 3208 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Geometries, frequencies and total energies of the ab initio calculations.. See DOI: 10.1039/b602412b |
| This journal is © the Owner Societies 2006 |