FTIR spectroscopic and computational studies on hydrogen adsorption on the zeolite Li–FER
P. Nachtigalla, E. Garroneb, G. Turnes Palominoc, M. Rodríguez Delgadoc, D. Nachtigallováa and C. Otero Areán*c
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic and Centre for Biomolecules and Complex Molecular Systems, Flemingovo n. 2, CZ-16610, Prague, Czech Republic
bDipartimento di Scienza dei Materiali ed Ingegneria Chimica, Politecnico di Torino, I-10126, Turin, Italy
cDepartamento de Química, Universidad de las Islas Baleares, E-07122, Palma de Mallorca, Spain. E-mail: dqueep0@uib.es; Fax: +34 971 173426; Tel: +34 971 173251
First published on 5th April 2006
Abstract
The interaction, at a low temperature, between molecular hydrogen and the zeolite Li–FER was studied by means of variable temperature infrared spectroscopy and theoretical calculations using a periodic DFT model. The adsorbed dihydrogen molecule becomes infrared active, giving a characteristic IR absorption band (H–H stretching) at 4090 cm−1. Three different Li+ site types with respect to H2 adsorption were found in the zeolite, two of which adsorb H2. Calculations showed a similar interaction energy for these two sites, which was found to agree with the experimentally determined value of standard adsorption enthalpy of ΔH0 = −4.1 (±0.8) kJ mol−1. The results are discussed in the broader context of previously reported data for H2 adsorption on Na–FER and K–FER.
1. Introduction
Studies of reversible gas adsorption on zeolites and related microporous materials are relevant to several technological processes, including gas separation (based on differential adsorption of different gases), gas sensing and storage. Hence, a detailed understanding of gas–solid interactions and quantification of the interaction energy is of interest, not only from the point of view of fundamental research, but also because practical applications would benefit from such studies. However, reversible gas adsorption involves only weak gas–solid interactions, typically within the range of 2–20 kJ mol−1, which pose demanding requirements on both experimental measurements and theoretical calculations.On the experimental side, it was recently shown1,2 that variable temperature infrared spectroscopy is a convenient method to determine gas adsorption enthalpy when dealing with weak gas–solid interactions, and this method is applied here to the study of hydrogen adsorption (at low temperature) on the zeolite Li–FER. Detailed analysis of experimental data can be accomplished by means of a parallel theoretical investigation on the zeolite adsorption sites, the geometry of the adsorption complex, and the corresponding interaction energy. For this purpose, we have used a periodic density functional description which, by paying due consideration to the zeolite framework topology, gives a far more realistic description of the gas–solid interaction than that which can be obtained with simple cluster models.
Related studies on hydrogen adsorption at low temperature on Na–FER and K–FER were reported recently,3 so the results obtained for the H2–Li–FER system can be analysed in a broader context. By doing so, further insight into the gas adsorption process can be gained, which constitutes an added aim of the present work. It should also be noted that hydrogen interactions with cations in mordenite and chabazite were recently studied by means of theoretical calculations by Benco et al.4 and Solans-Monfort et al.5 Detailed calculations for H2 interactions with bare alkali metal cations were reported by Vitillo et al.6
2. Materials and methods
2.1. Experimental
The ferrierite sample used in this study was supplied by the Research Institute of Inorganic Chemistry, Ústí nad Labem. It was in the ammonium form and had a nominal Si ∶ Al ratio of 8.5 ∶ 1. From the parent zeolite, the lithium-exchanged sample was obtained by repeated ion exchange with a 0.5 M aqueous solution of LiCl. Powder X-ray diffraction of the exchanged sample showed good crystallinity, and all diffraction lines present in the diffractogram corresponded to the FER structure type. Complete ion exchange was checked by the absence of IR absorption bands corresponding to either the ammonium ion or the Brønsted acid Si(OH)Al group, which would be generated during thermal activation (see below) of the zeolite sample if total exchange of the alkali metal ion for ammonium did not take place in the parent NH4–FER material.For IR spectroscopic measurements, a thin self-supported wafer of the zeolite sample was prepared and activated (outgassed) in a dynamic vacuum (residual pressure <10−4 Torr) for 3 h at 700 K inside an IR cell,7 which allowed in situ sample activation, gas dosage and variable temperature IR spectroscopy to be carried out. After running the blank spectrum of the zeolite wafer at 77 K, the cell was dosed with hydrogen, it was then closed and IR spectra were recorded at several fixed temperatures within the range of 79–150 K, while simultaneously registering sample temperature and hydrogen equilibrium pressure inside the cell. A platinum resistance thermometer and a capacitance pressure gauge were used for that purpose. The precision of these measurements was about ±2 K and ±2 × 10−2 Torr, respectively. Transmission FTIR spectra were recorded at 3 cm−1 resolution using a Bruker IFS66 spectrometer. In order to check reproducibility, and also to improve accuracy, after completing a first series of measurements the infrared cell was outgassed, dosed again with hydrogen, and a second series of spectra was recorded.
2.2. Calculations
The orthorhombic unit cell of ferrierite (Immm space group) contains 36 T atoms and 72 O atoms. The equilibrium volume of the all-silica FER unit cell fitted previously (cell parameters a = 19.1468, b = 14.3040 and c = 7.5763 Å, volume 2076.70 Å3)8 was used for all calculations on H2–Li–FER. One framework Si atom was replaced by aluminium and the charge was compensated for by a Li+ cation. Calculations of H2 adsorption enthalpies were carried out for Li+ in the vicinity of the Al atom in the T1, T2, and T3 positions (numbering scheme of ref. 9). Since substitution of one aluminium for one silicon atom only causes a negligible change in the unit cell volume, the geometries of Li–FER and H2–Li–FER were optimized within the constraint of the unit cell parameters while all other degrees of freedom were fully relaxed.Calculations were performed using the periodic DFT method, employing the Perdew–Burke–Erzerhofer (PBE) exchange–correlation functional10,11 and the projector augmented wave approximation (PAW) of Blöchl, as adapted by Kresse and Joubert.12,13 The plane wave basis set with a kinetic energy cut-off of 400 eV was used. Brillouin-zone sampling was restricted to the gamma-point. Calculations were performed using the VASP program.14–17
Experimental determination of ΔH0 was carried out at temperatures around 100 K. To compare the calculated interaction energy (electronic interaction energy) with the experimental adsorption enthalpy value, it is necessary to include several corrections: the effect of ZPE, a thermal contribution, and the pV term. Zero-point energy (ZPE) corrections were calculated within the harmonic approximation. The effect of temperature (ΔU0(100) – ΔU0(0)) was evaluated from the partition function considering six degrees of freedom (see ref. 3 for details). Thermal excitation of vibrational degrees of freedom was considered. The ideal gas equation was used for the transition from internal energy change to enthalpy change (ΔH0(T) = ΔU0(T) + pV = ΔU0(T) + RT).
The correction to the electronic interaction energy (calculated with the periodic DFT model) was evaluated for the 1-T cluster model (Al(OH)4−M+ cluster), and this correction (ΔH0(100 K) − ΔEel(0 K) = 4.9 kJ mol−1) was used for all H2 adsorption sites considered in Li–FER. A detailed description of this approach can be found in ref. 3. Calculations on cluster models were performed with the PBE exchange–correlation functional and with the aug-cc-pVTZ basis set.18,19 Calculations with the atom-centered basis set were carried out with the Gaussian03 program suite.20C2v symmetry constraints were applied in all calculations with cluster models and interaction energies were corrected for the basis set superposition error.21 For comparison, the Li+ bare metal cation (0-T model) was also considered.
3. Results
3.1. Variable temperature FTIR spectroscopy
Representative variable temperature FTIR spectra (in the H–H stretching region) of H2 adsorbed on Li–FER are depicted in Fig. 1. A single H–H stretching band is seen, centred at 4090 cm−1. The bathochromic frequency shift, from the gas phase value (4163 cm−1) of the Raman-active H–H stretching vibration of the free H2 molecule, amounts to −73 cm−1 for Li–FER. For comparison, H2 adsorbed (at liquid nitrogen temperature) on the MFI-type zeolite Li–ZSM-5 was reported to give a single infrared absorption band at 4092 cm−1.22,23 | ||
| Fig. 1 Representative variable temperature FTIR spectra (zeolite blank subtracted) of H2 adsorbed on Li–FER. Temperature, in K, and pressure (Torr, in brackets) as shown. For clarity the spectra have been offset on the vertical scale. | ||
From the integrated intensity of variable temperature IR spectra, and by simultaneously measuring temperature and hydrogen equilibrium pressure, the standard adsorption enthalpy (ΔH0) and entropy (ΔS0) involved in the adsorption process were determined by using the VTIR method described in detail elsewhere.1 Briefly, at any given temperature, the integrated intensity of the IR absorption band should be proportional to surface coverage, θ, thus giving information on the activity (in the thermodynamic sense) of both the adsorbed species and the empty adsorbing sites; simultaneously, the equilibrium pressure does the same for the gas phase. Hence, the corresponding adsorption equilibrium constant, k, can be determined, and the variation of k with temperature leads to the corresponding values of adsorption enthalpy and entropy. For deriving these values, integrated band intensity, A, temperature, T, and hydrogen equilibrium pressure, p, were considered to be interrelated by the Langmuir-type equation
| θ = A/AM = k(T) p/[1 + k(T) p] | (1) |
| k(T) = exp[ΔS0/R] exp[−ΔH0/RT] | (2) |
| ln[A/(AM − A)p] = (−ΔH0/RT) + (ΔS0/R), | (3) |
The plot of the left-hand side of eqn (3)versus reciprocal temperature for all of the experimental measurements, depicted in Fig. 2, shows a good linear fit of eqn (3). Note that the value of AM needed (for which only an approximate estimation was known, since experimental points in Fig. 1 correspond to coverages far from saturation) was chosen as that giving the best linear fit of eqn (3) for all experimentally determined points (see ref. 1 for details). From this AM value, it was inferred that experimental points in Fig. 2 correspond to a coverage range of 0.20 ≤ θ ≤ 0.65. From the linear plot in Fig. 2, the standard adsorption enthalpy was determined to be ΔH0 = −4.1 kJ mol−1 and the corresponding entropy change (ΔS0) is −57 J mol−1 K−1. The estimated error limits are about ±0.8 kJ mol−1 for the enthalpy and ±10 J mol−1 K−1 for the entropy.
 | ||
| Fig. 2 Plot of the left-hand side of eqn (3)versus reciprocal temperature for spectra measured in two independent runs (circle and square symbols). R, linear regression coefficient; and SD, standard deviation. | ||
3.2. Computational study
The structure of cation sites and coordination of the Li+ ion in FER was investigated systematically, considering all possible coordinations of the metal cation in the vicinity of each of four distinguishable framework aluminium atoms.24 Two site types were found in the vicinity of a single framework Al atom: (i) channel wall site (denoted M7/T3) where Li+ is located on top of a 6-membered ring on the channel wall, and (ii) intersection site (I2/T2) where Li+ is located in the 8-membered entrance window of the perpendicular channel, at the intersection of the main and perpendicular channels. The interaction of H2 with Li+ cations in the zeolite was studied for the most stable Li+ site of each type, M7/T3 and I2/T2. In addition, H2 adsorption on the P6/T1 site, that has a rather peculiar geometry (the Li+ ion is located right in the plane of the 6-membered window located at the bottom of FER cage) was also studied. The investigated sites are depicted in Fig. 3.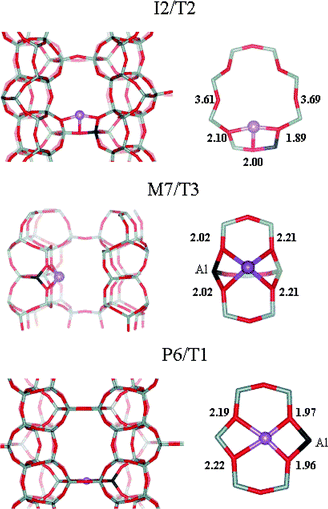 | ||
| Fig. 3 The most stable Li+ sites in FER. The localization of the site is depicted in the left part of the figure, viewed along the perpendicular, main, and perpendicular channel for the I2/T2, M7/T3, and P6/T1 site, respectively. The I2/T2 site is at the 8-membered entrance window of the perpendicular channel, on the intersection of both channels. The M7/T3 site is located in the main channel. The P6/T1 site is located in the plane of the 6-membered ring separating two adjacent FER cages. Details of Li+ coordination for each site type are given in the right-hand side. Distances between the Li+ ion and closest framework oxygen atoms are shown in Å. | ||
Results obtained with the periodic DFT model (FER) are reported in Table 1, along with those obtained using less sophisticated 0-T and 1-T cluster models. The strongest interaction (−23.5 kJ mol−1) was found for H2 adsorption on a bare Li+ cation (0-T model). This is not surprising in light of previous theoretical work5 which showed that the H2 interaction with alkali metal cations is dominated by the polarization contribution. The interaction of H2 with the Li+ site represented by the 1-T model is five times weaker than interaction with the bare Li+ cation. Calculations performed on the periodic model gave ΔH0 values which are much smaller than that for the bare cation (as expected), but they are comparable to, or even larger than, the adsorption enthalpy obtained with the 1-T cluster model.
| H2 Locationb | r(H–H)/Å | r(Li–H)/Å | ΔH0(100)/kJ mol−1 | |
|---|---|---|---|---|
| a Alkali metal sites are defined in Fig. 1.b M and P denotes the H2 molecule located in the main and in the perpendicular channel of FER, respectively.c Adsorption complex of two H2 molecules on the I2/T2 site depicted in Fig. 4a.d Average adsorption enthalpy.e A positive value of ΔH0(100) results from the use of constant correction (ΔH0(100 K) − ΔEel(0 K) = 4.9 kJ mol−1) for all sites. | ||||
| Cluster model | ||||
| 0-T | 0.7609 | 2.026 | −23.5 | |
| 1-T | 0.7547 | 2.155 | −4.7 | |
| FERa | ||||
| M7/T3 + H2 | M | 0.7549 | 2.180, 2.204 | −4.6 |
| I2/T2 + H2 | M | 0.7559 | 2.206, 2.235 | −5.6 |
| I2/T2 + H2 | P | 0.7561 | 2.183, 2.195 | −5.9 |
| I2/T2 + 2 × H2cd | M | 0.7554 | 2.207, 2.242 | −5.2 |
| P | 0.7561 | 2.220, 2.229 | ||
| P6/T1 + H2e | P | 0.7536 | 2.479, 2.434 | 0.1 |
| Experimental value | −4.1 (±0.8) | |||
The values of H2 adsorption enthalpy on the Li+ cation at the intersection site (I2/T2) were −5.6 and −5.9 kJ mol−1 for the H2 molecule approaching the Li+ ion from the main and the perpendicular channel, respectively. It is apparent that two H2 molecules can interact with one Li+ at the intersection site (Fig. 4a) at the same time without affecting each other. The H2 adsorption enthalpy on the Li+ cation at the channel wall site (M7/T3) is slightly smaller (by about 1 kJ mol−1) than that for the intersection site. However, H2 cannot be adsorbed on the Li+ ion at the P6/T1 site.
 | ||
| Fig. 4 Adsorption complexes of two H2 molecules on (a) Li+ and (b) Na+ cations at the I2/T2 intersection site (viewed along the main channel). The nearest framework oxygen atoms are on average 2.91 and 3.29 Å away from hydrogen atoms in Li–FER and Na–FER zeolites, respectively. | ||
For all models investigated (0-T, 1-T, and periodic model), the structures having the lowest energy were T-shaped adsorption complexes. The H–H bond length is elongated by only a few thousandths of an Å upon interaction with the alkali metal cation. (Note that in the gas phase H2 molecule r(H–H) is 0.751 Å at the PBE level using both the aug-cc-pVTZ and plane-wave basis sets). The largest H–H bond length elongation is found for H2 interacting with a bare Li+ ion (almost 0.01 Å), the smallest change in H–H bond length is observed for the least stable P6/T1 site. It is apparent from Table 1 that the calculated ΔH0 values correlate with corresponding H–H bond elongation. The coordination of the metal cation to the zeolite framework was found to be unaltered upon interaction with adsorbed H2.
4. Discussion
The experimentally determined value of ΔH0 = −4.1 kJ mol−1 is close to the corresponding values calculated using the periodic DFT approach (Table 1), excluding site P6/T1 which should not contribute to hydrogen adsorption. It should be noted, however, that the excellent agreement found between experimental and calculated values of standard adsorption enthalpy could, to some extent, be fortuitous. Two main sources of uncertainty can affect calculated ΔH0 values. First, the interaction energy calculated using GGA functionals (including the PBE functional) is known to be somewhat overestimated, while attraction due to dispersion forces is neglected at the DFT level, so these two terms can partially cancel out calculation errors. Secondly, the thermal contribution to ΔU0(100) was calculated from the vibrational partition function of the adsorption complex, using frequency values obtained at the harmonic approximation for the 1-T cluster model, and it is worth noticing that this approximation can be rather inaccurate, particularly for low lying vibrational modes. In addition, use of the 1-T cluster model neglects interaction of the H2 molecule with the zeolite channel wall. More refined calculations, however, would require a degree of complexity which is well beyond the scope of the present work.Regarding multiplicity of adsorption sites, theoretical calculations have clearly shown that the H2 molecule can be adsorbed on two different site types: the intersection site (I2/T2) and channel wall site (M7/T3). In addition, two different adsorption complexes (having either one or two adsorbed molecules) can be formed on the intersection site. By contrast, IR spectra show only a single infrared absorption band (Fig. 1) and, in addition to that, the experimental data could be interpreted by assuming a single (homogeneous) adsorbing site, i.e., within the context of an ideal Langmuir behaviour, which is implicit in eqn (3). These seemingly contrasting results can be reconciled by noticing that differences in the interaction energy for individual adsorption sites are not greater than 1.3 kJ mol−1, so (i) these differences are too small to cause a significant deviation from the ideal Langmuir behaviour, and (ii) IR spectroscopy (at 3 cm−1 resolution) cannot resolve infrared absorption bands originating from the different adsorption complexes formed in the H2–Li–FER system.
As already mentioned in the Introduction, a comparison of results obtained in this study with previously reported data3 for the H2–Na–FER and H2–K–FER systems is of interest. Table 2 summarizes experimentally determined values for the three gas–solid systems. The bathochromic frequency shift, Δν, is seen to follow a homogeneous trend (as expected), being largest for H2 adsorbed on Li–FER and smallest for K–FER. However, the adsorption enthalpy, ΔH0, does not follow the same trend: for the H2–Li–FER system, the corresponding value (−4.1 kJ mol−1) is lower than that found for the H2–Na–FER system (−6.0 kJ mol−1). This somewhat puzzling observation finds a precedent in previously reported studies22,25 on H2 adsorption on alkali metal exchanged ZSM-5 zeolites. For H2–Li–ZSM-5, reported values of Δν and ΔH0 are −71 cm−1 and −6.5 kJ mol−1, respectively, while for H2–Na–ZSM-5 they are −62 cm−1 and −10.3 kJ mol−1. It should also be noted that, in agreement with experimental results, the calculated adsorption enthalpy of H2 on Li–FER (−4.6 to −5.9 kJ mol−1, Table 1) is smaller than that found3 for H2 on Na–FER (−5.2 to −7.0 kJ mol−1). A tentative explanation for the H2–Li–ZSM-5 system showing a larger Δν value than H2–Na–ZSM-5, while ΔH0 is smaller for H2–Li–ZSM-5 than for H2–Na–ZSM-5, was given in ref. 22. Some of us proposed that, upon coordination to the adsorbed H2 molecule, the Li+ ion could slightly move outward from its original position on the zeolite wall, so as to optimize the interaction with the adsorbed molecule. Since this step would (necessarily) be endothermic, the overall value of ΔH0 would be lower than expected as compared to the H2–Na–ZSM-5 system. However, no theoretical calculations were performed at the time to substantiate this tentative hypothesis. We now report calculations for alkali metal exchanged ferrierite which give some evidence that cation movement is not a major reason for a low value of the adsorption enthalpy of H2 on Li+ sites.
The calculated energy terms and geometric parameters of Li+, Na+ and K+ interaction with the framework and the adsorbed H2 molecule are summarized in Table 3. Results obtained for the I2/T2 site are shown, together (for comparison) with corresponding values for the 1-T cluster model. Note that the H2 electronic interaction energy (ΔEel) calculated with the 1-T cluster model decreases monotonically from Li+ to K+, as expected from simple electrostatic considerations. However, at the periodic DFT level the electronic interaction energy is the same for H2 adsorbed on Li+ and Na+ sites. Upon accounting for the effect of the vibrational zero-point energy and thermal corrections, the H2 interaction with the Na+ site becomes stronger than that for the Li+ site, while the H2 interaction with the K+ site is the weakest.
| Li+ | Na+ | K+ | ||||
|---|---|---|---|---|---|---|
| 1-T | I2/T2 | 1-T | I2/T2 | 1-T | I2/T2 | |
| a Electronic interaction energy.b H2 adsorption enthalpy.c Zeolite deformation energy is defined as the energy difference between the M–FER system energy at the geometry of the H2–M–FER adsorption complex and at the equilibrium geometry of bare M-FER.d Average change of metal oxygen distance upon the interaction with H2.e Distance between the metal cation and adjacent framework oxygen atoms, distance to O atom that is not part of AlO4 tetrahedron in italic. | ||||||
| ΔEela/kJ mol−1 | −9.6 | −10.8 | −6.9 | −10.8 | −2.6 | −5.2 |
| ΔH0 (100)b/kJ mol−1 | −4.7 | −5.9 | −3.2 | −7.0 | −0.2 | −2.8 |
| ΔEdeformc/kJ mol−1 | 0 | 0.9 | 0 | 0.1 | 0 | 0.1 |
| Δr(M–O)d/Å | 0.01 | 0.02 | 0 | 0.01 | 0 | 0 |
| r(M–H)/Å | 2.16 | 2.19 | 2.58 | 2.45 | 3.20 | 3.15 |
| r(M–O)e/Å | 1.79 | 1.91 | 2.12 | 2.26 | 2.48 | 2.87 |
| 1.79 | 2.01 | 2.12 | 2.48 | 2.48 | 2.90 | |
| 2.17 | 2.49 | 3.23 | ||||
| 3.29 | ||||||
In order to examine whether the suprisingly small H2 adsorption enthalpy on Li–FER is due to a cation displacement (leading to an endothermic step), the geometrical changes and corresponding framework deformation energy are also reported in Table 3. It is clear that there are only very small changes in the cation to framework oxygen distance (0.02 and 0.01 Å for Li–FER and Na–FER, respectively). In addition, the framework deformation energy (defined as the difference between the M–FER system energy at the geometry of the H2–M–FER adsorption complex and at the equilibrium geometry of bare M-FER) is only 0.9 and 0.1 kJ mol−1 for Li–FER and Na–FER, respectively. This suggests that the endothermic step is not contributing much to the overall adsorption enthalpy for these systems. However, it should be noted that for strongly adsorbed molecules, the framework deformation energy significantly lowers the adsorption enthalpy, as shown recently for CO and NO adsorption on Cu–ZSM-5.26,27
The Li+ ion is tightly coordinated to oxygen atoms of the zeolite framework, as shown in Fig. 4. The geometrical parameters of H2–M–FER complexes (M = Li and Na) are shown in Table 3. Note that going from the 1-T cluster model to the periodic model the M–H distance increases slightly in the Li+ complex, while for the Na+ complex it shortens by over 0.1 Å. In agreement with this observation, the calculated H2 adsorption enthalpy on Li+ and Na+ cations was found to increase by 0.9 kJ mol−1 (Table 1) and 3.8 (ref. 3) kJ mol−1, respectively, on going from the 1-T cluster model to the periodic FER model.
Fig. 4 depicts the adsorption complexes formed by two H2 molecules in Li–FER and Na–FER (I2/T2 site). For Li–FER the adsorbed H2 molecule comes significantly closer to adjacent framework oxygen atoms than for Na–FER. Average distances between H atoms and closest framework oxygens (shown by black segments in Fig. 4) are 2.91 Å for Li–FER and 3.29 Å for Na–FER. Calculations of pair-interaction energy between H2 and the zeolite framework (at the geometry of the corresponding adsorption complex) gave −0.7 and −1.9 kJ mol−1 for H2–Li–FER and for H2–Na–FER, respectively. Considering uncertainties inherent to the calculations performed, these energy values are too small to be confidently discussed. In addition, the H2 interaction with the framework for other Li+ and Na+ sites in FER was found to be even smaller. Therefore, the reason for the enthalpy of adsorption of H2 on Li–FER being smaller than that on Na–FER remains unclear. However, the above calculations suggest that, (i) deformation of the adsorption site upon H2 coordination to the Li+ ion is not the major factor involved, (ii) tight Li+ coordination to the zeolite framework lowers the Li+ ability to bind H2, and (iii) H2 interaction with the zeolite framework (at the equilibrium distance in the adsorption complex) could slightly stabilize the H2–Li–FER and H2–Na–FER complexes to different extents. Further studies seem to be desirable in order to shed light on these points.
5. Conclusions
The standard adsorption enthalpy of H2 on the zeolite Li–FER was experimentally determined to be ΔH0 = −4.1 (±0.8) kJ mol−1 by means of variable temperature IR spectroscopy. This value was found to agree within ±1.5 kJ mol−1 with that calculated by using a periodic DFT method. However, such a close agreement can (in part) be due to internal cancellation of errors in the theoretical calculations.Periodic DFT calculations showed that two different cation sites can adsorb hydrogen. One of them (M7/T3) is situated on the channel wall, the other (I2/T2) is at the intersection of two perpendicular channels. Only one H2 molecule can be adsorbed on the M7/T3 site, while two H2 molecules can be simultaneously adsorbed on the I2/T2 site. In both cases, the adsorption complex formed is T-shaped and the corresponding values of interaction energy are very similar. H2 cannot be adsorbed on the P6/T1 site, where the Li+ cation is located in the plane of a 6-membered ring separating adjacent FER cages.
Finally, it is worth noting that both DFT calculations and experimental results showed that the standard adsorption enthalpy of molecular hydrogen on Li–FER is about 1.5–2 kJ mol−1 smaller than that on Na–FER. By contrast, the bathochromic frequency shift (from the free molecule value of 4163 cm−1) of the adsorbed H2 molecule is larger for H2–Li–FER than for H2–Na–FER, with values of −73 and −63 cm−1, respectively.
Acknowledgements
The Spanish Ministry of Education and Science is gratefully acknowledged for supporting work done at the UIB (Project number: MAT2005-05350). Work in Prague was supported by Grants of ME CR No. LC512 and GA CR No. 203/06/0324. EG gratefully acknowledges funding from the Italian Ministry of Education (MIUR). Research Project FISR 2004.References
- E. Garrone and C. Otero Areán, Chem. Soc. Rev., 2005, 34, 846–857 RSC.
- C. Otero Areán, O. V. Manoilova, G. Turnes Palomino, M. Rodríguez Delgado, A. A. Tsyganenko, B. Bonelli and E. Garrone, Phys. Chem. Chem. Phys., 2002, 4, 5713–5715 RSC.
- C. Otero Areán, G. Turnes Palomino, E. Garrone, D. Nachtigallova and P. Nachtigall, J. Phys. Chem. B, 2006, 110, 395–402 CrossRef.
- L. Benco, T. Bucko, J. Hafner and H. Toulhoat, J. Phys. Chem. B, 2005, 109, 22491–22501 CrossRef CAS.
- X. Solans-Monfort, V. Branchadell, M. Sodupe, C. M. Zicovich-Wilson, E. Gribov, G. Spoto, C. Busco and P. Ugliengo, J. Phys. Chem. B, 2004, 108, 8278–8286 CrossRef CAS.
- J. G. Vitillo, A. Damin, A. Zecchina and G. Ricchiardi, J. Chem. Phys., 2005, 122, 114311 CrossRef.
- C. Otero Areán, O. V. Manoilova, A. A. Tsyganenko, G. Turnes Palomino, M. Peñarroya Mentruit, F. Geobaldo and E. Garrone, Eur. J. Inorg. Chem., 2001, 1739–1743 CrossRef.
- O. Bludsky, M. Silhan, P. Nachtigall, T. Bucko, L. Benco and J. Hafner, J. Phys. Chem. B, 2005, 109, 9631–9638 CrossRef CAS.
- P. A. Vaughan, Acta Crystallogr., 1966, 21, 983 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 48, 13115–13118 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B, 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, Gaussian Inc., Pittsburgh, PA. 2003 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553.
- G. Turnes Palomino, M. Rodríguez Delgado, N. M. Tsyganenko, A. A. Tsyganenko, E. Garrone, B. Bonelli, O. V. Manoilova and C. Otero Areán, Stud. Surf. Sci. Catal., 2005, 158, 853–860.
- C. Otero Areán, O. V. Manoilova, B. Bonelli, M. Rodríguez Delgado, G. Turnes Palomino and E. Garrone, Chem. Phys. Lett., 2003, 370, 631–635 CrossRef.
- R. Bulanek and P. Nachtigall, Appl. Catal., A, 2006 DOI:10.1016/j.apcata.2006.03.020.
- C. Otero Areán, M. Rodríguez Delgado, G. Turnes Palomino, M. Tomás Rubio, N. M. Tsyganenko, A. A. Tsyganenko and E. Garrone, Microporous Mesoporous Mater., 2005, 80, 247–252 CrossRef.
- M. Davidova, D. Nachtigallova, R. Bulanek and P. Nachtigall, J. Phys. Chem. B, 2003, 107, 2327–2332 CrossRef CAS.
- M. Davidova, D. Nachtigallova, P. Nachtigall and J. Sauer, J. Phys. Chem. B, 2004, 108, 13674–13682 CrossRef CAS.
| This journal is © the Owner Societies 2006 |