Air and water stable ionic liquids in physical chemistry
Frank Endres* and Sherif Zein El Abedin†
Faculty of Natural and Materials Sciences, Clausthal University of Technology, Robert-Koch-Str. 42, 38678, Clausthal-Zellerfeld, Germany. E-mail: frank.endres@tu-clausthal.de; Fax: 0049-5323-722460
First published on 17th March 2006
Abstract
Ionic liquids are defined today as liquids which solely consist of cations and anions and which by definition must have a melting point of 100 °C or below. Originating from electrochemistry in AlCl3 based liquids an enormous progress was made during the recent 10 years to synthesize ionic liquids that can be handled under ambient conditions, and today about 300 ionic liquids are already commercially available. Whereas the main interest is still focussed on organic and technical chemistry, various aspects of physical chemistry in ionic liquids are discussed now in literature. In this review article we give a short overview on physicochemical aspects of ionic liquids, such as physical properties of ionic liquids, nanoparticles, nanotubes, batteries, spectroscopy, thermodynamics and catalysis of/in ionic liquids. The focus is set on air and water stable ionic liquids as they will presumably dominate various fields of chemistry in future.
Preface
When one of us (FE) gave a lecture with the title “In situ STM investigations of metal electrodeposition in room temperature molten salts” on a Bunsenkolloquium in 1999 in Germany, one person from the audience asked how an STM can be operated in a molten salt at high temperatures. This question was unexpected at that time and the speaker (FE) answered as diplomatically as possible that these salts are liquid at room temperature, as mentioned in the title of the lecture. Nevertheless, this question made clear that such liquids were hardly known at that time in electrochemistry—not too surprising if one takes into account a worldwide output of maybe 50 papers per year in 1999 in the field of room temperature molten salts/ionic liquids. The lecture was commented as very interesting but unusual and a few people in the audience expressed an opinion that these liquids will never be employed in any technical process for the forthcoming 100 years. In the “Molten Salt Community” (maybe 20–30 groups worldwide) on the other hand, these “room temperature molten salts” were regarded as uncommon and as a curiosity for a while, maybe because they need more chemistry than simple metal halides. The experience of many colleagues working with these liquids showed that the expression “molten salt” has always been associated with “high temperature”, as we also had to learn. It was about in the middle of the 1990s when it was decided in the community to replace the term “room temperature molten salt” by “ionic liquid”, and an ionic liquid is defined today as a liquid consisting solely of cations and anions with a melting point of 100 °C and below. Although any high temperature molten salt is an ionic liquid, too, this novel term for the room temperature liquids clearly made a distinction, and we ourselves were never asked again how an in situ STM can be operated in molten salts at high temperatures.As we will show below, the output of papers with the expression “ionic liquid” started to increase about 2000, and in the following years even technical processes were introduced. The most famous one might be the BASIL-process from BASF (biphasic acid scavenging utilizing ionic liquids) where the side product of an organic reaction is an easy to process ionic liquid instead of a less favourable solid in the conventional process. Fortunately, it took only a few years since 1999 until the first commercial process was introduced.
In 2005 there were more than 1500 peer reviewed papers containing the expression “ionic liquid” or “ionic liquids”, and from 1995 to 2005 we found more than 4300 papers. About 30% of these papers deal with any aspect of physical chemistry. When we got the invitation to write this review article we had to make a selection, and we are well aware that other authors would probably have selected different topics. As we focus on several aspects of “interface electrochemistry” in our own research field we summarize more or less completely the state-of-the-art of nano-electrochemistry and electrodeposition in air and water stable ionic liquids. Furthermore, we introduce the physical properties of ionic liquids, nanoparticles, nanotubes, batteries, spectroscopy, thermodynamics and catalysis of/in ionic liquids. We focus on air and water stable ionic liquids, as in our opinion they will dominate various fields of chemistry in the future—not at all surprising if one takes into account that theoretically 1018 different ionic liquids are possible. The AlCl3-based ionic liquids, with which the research began seriously in the 1980s, will rather survive in electrochemistry, e.g. for the electrodeposition of aluminium and its alloys. We hope that with our review article an inexperienced reader will get a starting point to find his own way in the physical chemistry of ionic liquids.
1. Introduction
1.1 A brief history
The early history of ionic liquids began in 1914 when the first report of a room temperature molten salt was reported by Walden.1 He reported the physical properties of ethylammonium nitrate, [C2H5NH3] NO3, which has a melting point of 12 °C, formed by the reaction of ethylamine with concentrated nitric acid. Then, Hurley and Weir2 stated that a room temperature ionic liquid could be prepared by mixing and warming 1-ethylpyridinium chloride with aluminum chloride. In 1970s and 1980s, Osteryoung et al.3,4 and Hussey et al.5–7 carried out extensive research on organic chloride–aluminium chloride ambient temperature ionic liquids and the first major review of room temperature ionic liquids was written by Hussey.8 The ionic liquids based on AlCl3 can be regarded as the first generation of ionic liquids.The hygroscopic nature of AlCl3 based ionic liquids has delayed the progress in their use in many applications since they must be prepared and handled under inert gas atmosphere. Thus, the synthesis of air and water stable ionic liquids, which are considered as the second generation of ionic liquids, attracted further interest in the use of ionic liquids in various fields. In 1992, Wilkes and Zaworotko9 reported the first air and moisture stable ionic liquids based on 1-ethyl-3-methylimidazolium cation with either tetrafluoroborate or hexafluorophosphate as anions. Unlike the chloroaluminate ionic liquids, these ionic liquids could be prepared and safely stored outside of an inert atmosphere. Generally, these ionic liquids are water insensitive, however, the exposure to moisture for a long time can cause some changes in their physical and chemical properties. From our experience, we have found using in situ scanning tunneling microscopy that the undried ionic liquid [BMIm] PF6 attacks the gold substrate, and its aggressiveness increases with the increase in water content. This is due to the formation of HF as a result of decomposition of the ionic liquid in presence of water. Therefore, ionic liquids based on more hydrophobic anions such as tri-fluoromethanesulfonate (CF3SO−3), bis-(trifluoromethanesulfonyl) imide [(CF3SO2)2N−] and tris-(trifluoromethanesulfonyl) methide [(CF3SO2)3C−] have been developed.10–12 These ionic liquids have received extensive attention not only because of their low reactivity with water but also because of their large electrochemical windows. Usually these ionic liquids can be well dried the water contents below 1 ppm under vacuum at temperatures between 100 and 150 °C.
The histogram of Fig. 1 shows the increase of the number of publications on ionic liquids during the last decade up to now. As seen, the average number of publications in the last decade is about 40 papers per year while in 2004 about 1000 papers and in 2005 about 1500 papers were published. This reflects the increased interest in ionic liquids in general.
 | ||
| Fig. 1 Publications containing the phrase “ionic liquid or ionic liquids” in the title; abstract and key words; determined by ISI web of science; as a function of time. | ||
Beside Osteryoung, Wilkes, Hussey and Seddon who are pioneers in the field of ionic liquids, there are several scientists, e.g. Rogers, Welton, Wasserscheid, MacFarlane, Ohno, Endres, Davis, Jr, Abbott, and others, who entered this field having a strong impact in introducing the ionic liquids in many applications.
Rogers is one of the highly cited authors in the field of ionic liquids. He focuses on the synthesis and characterization of environmentally friendly ionic liquids as green solvents. He measured and published physicochemical properties data for many ionic liquids with the aim of providing data to start evaluating the use of ionic liquids in a variety of processes. Also, he works on the development of new materials from cellulose utilizing ionic liquids.
Welton has published many papers dealing with the applications of ionic liquids as solvents for synthesis and catalysis. He focuses on how the ionic liquids interact with solute species to affect their reactivity and he works on replacing environmentally damaging solvents with more benign alternatives. He is also the author of one of the most cited papers13 which was cited 1719 times up to November 2005.
Wasserscheid is an active member of the ionic liquid community and focuses on the preparation and characterization of ionic liquids for use in the biphasic catalysis. For example, he could show that the use of hexafluorophosphate ionic liquids allows selective, biphasic oligomerization of ethylene to 1-olefins. Together with Welton, he edited a very important book entitled Ionic Liquids in Synthesis which presents the synthesis and physicochemical properties of ionic liquids as well as their use in catalysis, polymerization, and organic and inorganic synthesis.14
MacFarlane works on the synthesis of new air and water stable ionic liquids with the purpose of employing such ionic liquids as indicators for sensing and displaying an environmental parameter such as humidity. This process is controlled by the colour change of the ionic liquids where they are synthesized with either a coloured cation or anion, so that the ionic liquids themselves are sensors. Also, he has published many papers on the use of ionic liquids in electropolymerization and in batteries.
Ohno concentrates his work on the synthesis of a series of polymerizable ionic liquids and their polymerization to prepare a new class of ion conductive polymers. For example, he prepared polymer electrolytes with high ionic conductivity and good elasticity by mixing nitrite rubber (poly(acrylonitrile-co-butadiene) rubber) with the ionic liquid N-ethylimidazolium bis(trifluoromethanesulfonyl)imide. Quite recently, he edited a book entitled Electrochemical aspects of ionic liquids which introduces some basic and advanced studies on ionic liquids in the field of electrochemistry.15
Davis, Jr introduced the concept of “task-specific ionic liquids” (TSILs) in the field of ionic liquids. TSILs are ionic liquids in which a functional group is incorporated enabling the liquid to behave not only as a reaction medium but also as a reagent or catalyst in some reactions or processes.
Abbott has recently developed a range of ionic compounds, which are fluid at room temperature. These ionic liquids are based on simple precursors such as choline chloride (vitamin B4) which is cheap and produced on a multitonne scale and hence these ionic liquids/deep eutectic solvents can be applied to large scale processes for the first time. Using these liquids, a number of applications are now under development such as electrodeposition of metals, electropolishing and ore processing.
We ourselves (Endres and Zein El Abedin) started about 10 years ago to study nanoscale processes at the interface electrode/ionic liquid using in situ (electrochemical) scanning tunneling microscopy (in situ-STM). We could show for the first time that Ge, Si, Se, Ta and Al can be electrodeposited in high quality in air and water stable ionic liquids. Presumably many more elements and compounds can be made electrochemically. Some recent results of the nanoscale electrodeposition in water and air stable ionic liquids will be presented.
2. Physical properties of ILs
2.1. Conductivity
Ionic liquids have reasonably good ionic conductivities compared with those of organic solvents/electrolyte systems (up to ∼10 mS cm−1).16 At elevated temperatures of e.g. 200 °C a conductivity of 0.1 Ω−1 cm−1 can be achieved for some systems. However, at room temperature their conductivities are usually lower than those of concentrated aqueous electrolytes. Based on the fact that ionic liquids are composed solely of ions, it would be expected that ionic liquids have high conductivities. This is not the case since the conductivity of any solution depends not only on the number of charge carriers but also on their mobility. The large constituent ions of ionic liquids reduce the ion mobility which, in turn, leads to lower conductivities. Furthermore, ion pair formation and/or ion aggregation lead to reduced conductivity. The conductivity of ionic liquids is inversely linked to their viscosity. Hence, ionic liquids of higher viscosity exhibit lower conductivity. Increasing the temperature increases conductivity and lowers viscosity.2.2. Viscosity
Generally, ionic liquids are more viscous than common molecular solvents and their viscosities are ranging from 10 mPa s to about 500 mPa s at room temperature. The viscosities of some popular air and water stable ionic liquids at room temperature are: 312 mPa s for [BMIm]PF6;17 154 mPa s for [BMIm]BF4;18 52 mPa s for [BMIm]TF2N;10 85 mPa s for [BMP]TF2N.12 The viscosity of ionic liquids is determined by van der Waals forces and hydrogen bonding. Electrostatic forces may also play an important role. Alkyl chain lengthening in the cation leads to an increase in viscosity.10 This is due to stronger van der Waals forces between cations leading to increase in the energy required for molecular motion. Also, the ability of anions to form hydrogen bonding has a pronounced effect on viscosity. The fluorinated anions such as BF−4 and PF−6 form viscous ionic liquids due to the formation of hydrogen bonding.19 In general, all ionic liquids show a significant decrease in viscosity as the temperature increases (see, e.g., ref. 20).2.3. Density
Ionic liquids in general are denser than water with values ranging from 1 to 1.6 g cm−3 and their densities decrease with increase in the length of the alkyl chain in the cation.21 For example, in ionic liquids composed of substituted imidazolium cations and CF3SO3− anion the density decreases from 1.39 g cm−3 for [EMIm]+ to 1.33 g cm−3 for [EEIm]+, to 1.29 g cm−3 for [BMIm]+ and to 1.27 g cm−3 for [BEIm]+.22 The densities of ionic liquids are also affected by the identity of anions. For example, the densities of 1-butyl-3-methylimidazolium type ionic liquids with different anions, such as BF4, PF6, TFA and Tf2N are 1.12 g cm−3,23 1.21 g cm−3,10 1.36 g cm−323 and 1.43 g cm−3,10 respectively. The order of increasing density for ionic liquids composed of a single cation is: [CH3SO3]− ≈ [BF4]− < [CF3CO2]− < [CF3SO3]− < [C3F7CO2]− < [(CF3SO2)2N]−.222.4. Melting point
As a class, ionic liquids have been defined to have melting points below 100 °C and most of them are liquid at room temperature. Both cations and anions contribute to the low meting points of ionic liquids. The increase in anion size leads to a decrease in melting point.24 For example, the melting points of 1-ethyl-3-methylimidazolium type ionic liquids with different anions, such as [BF4]− and [Tf2N]− are 15 °C25 and −3 °C,10 respectively. Cations size and symmetry make an important impact on the melting points of ionic liquids. Large cations and increased asymmetric substitution results in a melting point reduction.262.5. Thermal stability
Ionic liquids can be thermally stable up to temperatures of 450 °C. The thermal stability of ionic liquids is limited by the strength of their heteroatom–carbon and their heteroatom–hydrogen bonds, respectively.24 Wilkes et al.27 reported that the ionic liquids 1-ethyl-3-methyl-imidazolium tetrafluoroborate, 1-butyl-3-methyl-imdazolium tetrafluoroborate and 1,2-dimethyl-3-propyl imidazolium bis(trifluorosulfonyl)imide are stable up to temperatures of 445, 423 and 457 °C, respectively. Our experiences shows that such high temperatures are only tolerated by most liquids for a short time. Long time exposure to such high temperatures inevitably leads to decomposition. Most of the ionic liquids have extremely low vapour pressures, which allows to remove water by simple heating under vacuum. Water contents below 1 ppm are quite easy to achieve with most of the liquids.2.6. Electrochemical window
The electrochemical window is an important property and plays a key role in using ionic liquids in electrodeposition of metals and semiconductors. By definition, the electrochemical window is the electrochemical potential range over which the electrolyte is neither reduced nor oxidized at an electrode. This value determines the electrochemical stability of solvents. As known, the electrodeposition of elements and compounds in water is limited by its low electrochemical window of only about 1.2 V. On the contrary, ionic liquids have significantly larger electrochemical windows, e.g., 4.15 V for [BMIm]PF6 at a platinum electrode,28 4.10 V for [BMIm]BF428 and 5.5 V for [BMP]Tf2N at a glassy carbon electrode.12 In general, the wide electrochemical windows of ionic liquids have opened the door to electrodeposit metals and semiconductors at room temperature which were formerly obtained only from high temperature molten salts. For example, Al, Mg, Si, Ge, and rare earth elements can be obtained from room temperature ionic liquids. The thermal stability of ionic liquids allows to electrodeposit Ta, Nb, V, Se and presumably many other ones at elevated temperature.3. Electrosynthesis in air and water stable ionic liquids
In this section we will report on the use of some popular air and water stable ionic liquids such as, ZnCl2/[EMIm] Cl, [EMIm] BF4, [BMIm] BF4, [BMIm] PF6, [BMP] Tf2N, [BMIm] Tf2N and choline chloride–MCl in electrodeposition of metals and semiconductors in the bulk phase. Furthermore we will introduce nanoscale processes at the interface electrode/ionic liquid as well as in electropolymerization. We focus solely on the novel air and water stable liquids, as in our opinion they will be of significant interest for several aspects of electrochemistry.3.1. Electrodeposition of metals and alloys
Katayama et al.29 have reported that a room temperature ionic liquid 1-ethyl-3-methylimidazolium tetrafluoroborate ([EMIm]BF4) is applicable as an alternative electroplating bath for silver. The ionic liquid [EMIm]BF4 is superior to the chloroaluminate systems since the electrodeposition of silver can be performed without the risk of aluminium codeposition. Electrodeposition of silver in the ionic liquids 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIm]BF4) and [BMIm]PF6 was also reported in ref. 30. It was furthermore stated that Cd,31 Cu32 and Sb33 can be electrodeposited in a mixture of 1-ethyl-3-methyl imidazolium tetrafluoroborate ([EMIm]BF4) and [EMIm]Cl. Recently, Sun et al. have demonstrated that compound semiconductors such as indium antimonide (InSb)34 and cadmium telluride (CdTe)35 can be electrodeposited in the Lewis basic 1-ethyl-3-methylimidazolium tetrafluoroborate ionic liquid [BMIm]BF4. InSb is a III–V compound semiconductor and CdTe is a II–VI semiconductor, both are widely used in many fields such as electronic devices and solar cells.It was stated in ref. 36 that titanium can be electrodeposited in thin layers of maybe 5 nm at room temperature in the ionic liquid 1-butyl-3-methylimidazolium bis (trifluoromethylsulfonyl) imide [BMIm] Tf2N. With all refractory metals the challenge in depositing micrometer thick solely metallic layers is to avoid the growth of non-stoichiometric subhalides.
It has been shown that ionic liquids can be formed by the combination of zinc chloride with pyridinium-,37 dimethylethylphenyl-ammonium-,38 1-ethyl-3-methylimidazolium chloride [EMIm]Cl and 1-butyl-3-methylimidazolium chloride [BMIm]Cl.39–41 These ionic liquids are quite easy to prepare and do not decompose in the presence of water and air. It was reported42 that the potential limits for a basic 1 ∶ 3 ZnCl2–[EMIm]Cl ionic liquid corresponds to the cathodic reduction of [EMIm]+ and anodic oxidation of Cl−, giving an electrochemical window of approximately 3.0 V. For acidic ionic liquids that have a ZnCl2–[EMIm]Cl molar ratio higher than 0.5 ∶ 1, the negative potential limit is due to the deposition of metallic zinc, and the positive potential limit is due to the oxidation of the chlorozincate complexes. As a result of this fact, the electrodeposition of Zn and its alloys is possible in the Lewis acidic liquids. It was shown that Lewis acidic ZnCl2–[EMIm]Cl (in which the molar percentage of ZnCl2 is higher than 33 mol%) are potentially useful for the electrodeposition of zinc and zinc containing alloys.43–45 Huang and Sun have reported that Pt–Zn alloy,46 iron and Zn–Fe alloy,47 tin and Sn–Zn alloy,48 cadmium and Cd–Zn alloy49 can be electrodeposited in Lewis acidic ZnCl2–[EMIm]Cl ionic liquids.
Abbott et al.50 have reported the synthesis and characterization of new moisture stable, Lewis acidic ionic liquids/deep eutectic solvents made from metal chlorides and quaternary ammonium salts which are commercially available. They have shown that mixtures of choline (2-hydroxyethyltrimethylammonium) chloride [(H3C)3NC2H4OH)Cl] and MCl2 (M = Zn, Sn) give conducting and viscous liquids at or around room temperature. These liquids are easy to prepare, they are water and air insensitive and their low costs enable their use in large scale applications. Furthermore, they have reported51 that a dark green, viscous liquid can be formed by mixing choline chloride with chromium(III) chloride hexahydrate and the physical properties of this liquid are characteristic of an ionic liquid. The eutectic composition is found to be 1 ∶ 2 choline chloride/chromium chloride. From this ionic liquid chromium can be electrodeposited efficiently to yield a crack-free deposit.51 Addition of LiCl to the choline chloride/CrCl3·6H2O mixture was found to allow the deposition of nanocrystalline black chromium films.52 The use of this ionic liquid might offer an environmentally friendly process for electrodeposition of chromium instead of the currently used chromic acid based baths.
3.2. Electrodeposition on the nanoscale
Almost 10 years ago we started for the first time with in situ STM studies on electrochemical phase formation in ionic liquids. On the one hand, there was no knowledge on the local processes of phase formation in ionic liquids at all; on the other hand—due to wide electrochemical windows—these systems give access to elements that cannot be obtained in aqueous solutions, such as Al, Ge, Si, Ta and many more. Especially in the rapidly growing field of nanotechnology where semiconductor nanostructures will play an important role, we see a great chance for electrodeposition of nanostructures in ionic liquids. For this purpose the electrochemical processes and the factors that influence the deposition and the stability of the structures have to be understood on the nanometer scale.Fig. 2 shows the typical cyclic voltammogram of high purity and water free [BMIm]PF6 saturated with GeCl4. For a better comparison we have calibrated the processes vs. the germanium overpotential deposition that we observed in this system. As seen, we observe two main reduction peaks below the open circuit potential (OCP) and several oxidation peaks for electrode potentials above the OCP. The reduction peak at +500 mV vs. Ge corresponds to the reduction of Ge(IV) to Ge(II), the rising cathodic current at 0 V vs. Ge is correlated with the electrodeposition of elemental germanium that can even be seen with the naked eye as a black deposit formed on the electrode surface. The oxidation peak at +1000 mV is clearly correlated with Ge electrooxidation whereas the peaks above +1500 mV are also observed if the CV is cycled between +1000 and +3000 mV vs. Ge. These redox processes are correlated with the electrooxidation of the gold substrate.
 | ||
| Fig. 2 Cyclic voltammogram and a set of STM pictures recorded simultaneously on Au(111) in the ionic liquid 1-butyl-3-methylimidazolium-hexafluorophosphate, saturated with GeCl4. | ||
Fig. 2 shows furthermore a series of STM pictures where, together with the STM scan (from top to bottom) a cyclic voltammogram was run on Au(111) from GeCl4 saturated in [BMIm]PF6 with a scan rate of 10 mV s−1. Fig. 2a shows a typical Au(111) surface at +1200 mV vs. Ge. It is characterized by 250 pm high gold terraces and some gold islands. The electrode potential at the top of the STM picture of Fig. 2b is +1000 mV vs. Ge, it is 0 V at the bottom: it is quite evident that islands grow on the gold surface at potentials positive from the bulk deposition. Thus the deposition of Ge on Au(111) begins in the underpotential deposition regime. As we have pointed out in more detail in ref. 54, the underpotential deposition of Ge starts at the steps of the terraces at +1000 mV, then 150 pm high Ge islands start to be deposited at +950 mV and at about +750 mV vs. Ge we observe 250 pm high islands, presumably the result of alloying between Au and Ge. Still in the UPD regime a completely closed monolayer forms on the gold surface, as evidenced in Fig. 2b. The STM picture of Fig. 2c (taken within a potential range from 0 to −1000 mV, from top to bottom) shows the formation of a rough, thin Ge layer on gold surface. The thin Ge layer we obtain under these conditions can be ascribed to the relatively high potential scan rate which does not provide enough time for a massive bulk deposition. In the reverse scan, the rough Ge layer is stable in the potential range between −1000 and 0 mV, as revealed in the STM picture of Fig. 2d. In the potential range between 0 and +1000 mV, the Ge layer redissolves producing holes in the gold surface, which can be seen from a closer look at the STM picture of Fig. 2e (arrows in Fig. 2e). This is typical for surface alloying between deposit and substrate. On further potential scan (from +1200 to +2200 mV) gold oxidation occurs which starts first at the steps as can be clearly seen in the STM picture of Fig. 2f.
The STM pictures of Fig. 3 show the formation of triangularly-shaped Ge islands in [BMIm]PF6 saturated with GeBr4 as a source of germanium on Au(111). As seen in Fig. 3a, at a potential of +200 mV vs. Ge a rough and coherent layer of Ge is observed. In the upper left quarter of the picture a triangularly shaped island is imaged, its height is between 0.6 and 1 nm and does not seem to be “complete”. If the electrode potential is reduced to 0 V, islands of about 50–80 nm in diameter and with heights of up to 1.5 nm have formed and they start growing vertically very slowly, Fig. 3b. Upon further reducing the electrode potential the islands grow both vertically and laterally and finally merge.
 | ||
| Fig. 3 (a) “UPD” covered Au at +200 mV vs. Ge. The layer is coherent but rough. (b) Islands with heights of up to 1.5 nm form at 0 mV vs. Ge. (c) Height profile of the triangular islands. | ||
The current/voltage tunneling spectrum of an approximately 200 nm thick layer obtained at a potential of −250 mV vs. Ge is presented in Fig. 4. As can be seen in the original paper55 the surface is rough on the nanometer scale and some individual islands rise above the surface with heights of some nanometres. The I/U tunneling spectrum on such a Ge film shows a band gap of 0.7 ± 0.1 eV, in good agreement with the band gap of 0.67 eV for intrinsic microcrystalline bulk Ge at 300 K. In contrast to water, ionic liquids have due to their wide electrochemical window the benefit that no electrochemical side reactions like e.g. hydrogen evolution occur. Thus the band gap can be reliably measured in situ under electrochemical conditions—a challenge in aqueous solutions.
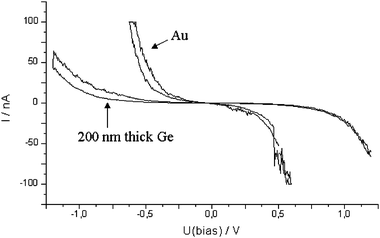 | ||
| Fig. 4 In situI–U tunneling spectra of an approximately 200 nm thick Ge layer and gold substrate. | ||
Not only Ge layers can be obtained but also Ge nanoclusters can be made by adjusting the experimental parameters. In ref. 56 we could show that with GeCl4 concentrations of about 5 × 10−3 mol l−1 narrowly distributed nanoclusters can be electrodeposited on Au(111). In situ current–voltage tunneling spectroscopy on 10 nm thick clusters has clearly shown a band gap of 0.7 ± 0.1 eV, and there seems to be a metal to semiconductor transition with increasing layer thickness.
![(1) Electrochemical window of [BMP]Tf2N on HOPG with the ferrocene/ferrocinium couple. (2) Cyclic voltammogram of SiCl4 saturated in the same ionic liquid. Scan rate each: 10 mV s−1.](/image/article/2006/CP/b600519p/b600519p-f5.gif) | ||
| Fig. 5 (1) Electrochemical window of [BMP]Tf2N on HOPG with the ferrocene/ferrocinium couple. (2) Cyclic voltammogram of SiCl4 saturated in the same ionic liquid. Scan rate each: 10 mV s−1. | ||
If the SiCl4 saturated ionic liquid is investigated, a strong reduction current sets in at an electrode potential which is 600 mV positive from the cathodic decomposition limit of the liquid on HOPG. After having passed the lower switching potential the anodic scan crosses the cathodic one at −2000 mV vs. Fc/Fc+ which is typical for nucleation. Approaching an electrode potential of +400 mV vs. Fc/Fc+ a strong oxidation current starts which is in part correlated to the SiCl4 reduction process beginning at −1600 mV vs. Fc/ Fc+ and in part correlated to HOPG oxidation as with SiCl4 in the liquid a similar oxidation behaviour is observed if the scan is started from the open circuit potential towards positive potentials.
Fig. 6 shows a high-resolution SEM picture of an electrodeposited silicon layer on gold substrate. As seen, the deposit contains small crystallites with sizes of around 50 nm. Often the deposit keeps its dark appearance even under air. The EDX analysis gave as a result only gold from the substrate and silicon, but no detectable chlorine. This proves that obviously elemental silicon was electrodeposited which is subject to some oxidation under environmental conditions.
 | ||
| Fig. 6 SEM micrograph of electrodeposited silicon, made potentiostatically at –2.7 V vs. Fc/Fc+. | ||
Fig. 7a shows the STM picture of an about 100 nm thick silicon layer that was electrodeposited at −1600 mV vs. Fc/Fc+, probed under potential control with the in situ STM. Its surface is smooth on the nanometer scale. Fig. 7b shows an in situ current/voltage tunneling spectrum of HOPG (curve 1) and of the 100 nm thick silicon layer (curve 2). The spectra are all over the surface of the same quality. Whereas the tunneling spectrum of HOPG is—as expected—metallic, for the silicon deposit a typical band gap is observed. An evaluation of the band gap gives a value of 1.0 ± 0.2 eV. This value is quite similar to the value that we observed for hydrogen terminated n-doped Si(111) in an ionic liquid.63 The value of microcrystalline silicon in the bulk phase at room temperature is 1.1 eV. In the light of these results, it can be concluded that elemental, intrinsic semiconducting silicon was electrodeposited from the employed ionic liquid.
 | ||
| Fig. 7 (a) In situ STM picture of an about 100 nm thick film (600 nm × 200 nm). (b) In situ current/voltage tunneling spectra of HOPG (curve 1) and of the silicon electrodeposit (curve 2) on HOPG. | ||
Fig. 8 shows the cyclic voltammogram of ([BMP]Tf2N) containing 0.5 M TaF5 on Au(111) at room temperature. As shown, two reduction processes are recorded in the forward scan. The first one starts at a potential of −0.5 V with a peak at −0.75 V, it might be correlated to the electrolytic reduction of Ta(V) to Ta(III). The second process starts at a potential of −1.5 V and is accompanied by the formation of a black deposit on the electrode surface. This can be attributed to the reduction of Ta(III) to Ta metal simultaneously with the formation of insoluble tantalum compounds. The anodic peak recorded on the backward scan is due to the dissolution of the electrodeposit which, however, is not completely reversible. At E > 1.5 V the anodic current increases as a result of gold dissolution. The deposit obtained only loosely adheres to the surface and it can easily be removed by washing with acetone.
![Cyclic voltammogram of 0.5 M TaF5 in ([BMP]Tf2N) on Au(111) at room temperature. Scan rate 10 mV s−1.](/image/article/2006/CP/b600519p/b600519p-f8.gif) | ||
| Fig. 8 Cyclic voltammogram of 0.5 M TaF5 in ([BMP]Tf2N) on Au(111) at room temperature. Scan rate 10 mV s−1. | ||
We also performed the electrodeposition of Ta at different temperatures of up to 200 °C. It was found that the mechanical quality and the adherence of the electrodeposits improve at 200 °C. Moreover, the quality and the adherence of the electrodeposit were found to be improved upon addition of LiF to the electrolyte.64 The SEM micrograph of the Ta electrodeposit (Fig. 9a) made potentiostatically at −1.8 V in ([BMP]Tf2N) containing 0.25 M TaF5 and 0.25 M LiF on Pt electrode at 200 °C for 1 h shows a smooth, coherent and dense layer. XRD patterns of the electrodeposit clearly show the characteristic patterns of crystalline tantalum, Fig. 9b.
![(a) SEM micrograph of the electrodeposit formed potentiostatically on Pt in ([BMP]Tf2N) containing 0.25 M TaF5 and 0.25 LiF at a potential of −1.8 V for 1 h at 200 °C. (b) XRD patterns of the deposited layer obtained potentiostatically on Pt in ([BMP]Tf2N) containing 0.25 M TaF5 and 0.25 LiF at a potential of −1.8 V for 1 h at 200 °C.](/image/article/2006/CP/b600519p/b600519p-f9.gif) | ||
| Fig. 9 (a) SEM micrograph of the electrodeposit formed potentiostatically on Pt in ([BMP]Tf2N) containing 0.25 M TaF5 and 0.25 LiF at a potential of −1.8 V for 1 h at 200 °C. (b) XRD patterns of the deposited layer obtained potentiostatically on Pt in ([BMP]Tf2N) containing 0.25 M TaF5 and 0.25 LiF at a potential of −1.8 V for 1 h at 200 °C. | ||
In situ STM measurements under potentiostatic conditions can give valuable information on the electrodeposition of Ta in the employed ionic liquid ([BMP]Tf2N). The STM picture of Fig. 10a shows a typical surface of gold on mica substrate (Au(111)) in the ionic liquid ([BMP]Tf2N) containing 0.5 M TaF5 at open circuit potential. As seen, the surface is characterized by terraces with average step heights of about 250 pm, typical for Au(111). By applying a potential of −1.25 V (vs. Pt) the nature of the surface changes, as seen in the STM picture of Fig. 10b. A rough layer of Ta forms rapidly and some triangularly shaped islands with heights of several nanometers grow above the deposited layer. With ongoing time, these islands grow vertically and laterally and finally merge together to a thick layer.
![(a) In situ STM picture of Au(111) in ([BMP]Tf2N) containing 0.5 M TaF5 at the open circuit potential (−0.2 V). (b) In situ STM picture of the electrodeposit obtained at a potential of −1.25 V.](/image/article/2006/CP/b600519p/b600519p-f10.gif) | ||
| Fig. 10 (a) In situ STM picture of Au(111) in ([BMP]Tf2N) containing 0.5 M TaF5 at the open circuit potential (−0.2 V). (b) In situ STM picture of the electrodeposit obtained at a potential of −1.25 V. | ||
The 3-D STM picture of Fig. 11a shows the topography of the electrodeposit, with a thickness of about 300 nm. In order to investigate if the in situ deposit is metallic or not, current/voltage tunneling spectroscopy was performed. A typical in situ tunneling spectrum of the 300 nm thick layer of the electrodeposit at different positions is shown in Fig. 11b. The I–U spectrum clearly exhibits metallic behaviour with an exponential-like rise of the current revealing that the electrodeposited layer might be elemental Ta. Together with the in situ measurements we can conclude that the reduction of TaF5 in ([BMP]Tf2N) leads to an at least 500 nm thick layer of metallic tantalum.
 | ||
| Fig. 11 (a) In situ 3-D STM picture of about 300 nm thick layer of the electrodepsoit. (b) In situI–U tunneling spectrum of the electrodeposit. | ||
3.3. Electrosynthesis of conducting polymers
Conducting polymers have attracted considerable attention as new materials for the development of numerous electrochemical devices such as batteries, supercapacitors, sensors, electrochromic devices, electrochemical actuators and light emitting diodes.65 These polymers can either be prepared by chemical or by electrochemical polymerization. The electrochemical synthesis offers some advantages, such as the generation of polymers in the doped state, the easy control of the film thickness. Furthermore, electropolymerization is an easy and rapid method.Recently attention has been directed to the potential benefits of using ionic liquids as solvents for the electrochemical synthesis of conducting polymers. Sekiguchi et al. reported the polymerisation of pyrrole, thiophene and aniline66,67 in 1-ethyl-3-methylimidazolium trifluoromethanesulfonate. MacFarlane and co-workers used the ionic liquids 1-butyl-3-methylimidazolium hexafluorophosphate, 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl) imide and 1-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl) imide, both as the growth medium and as an electrolyte for the electrochemical cycling of polypyrrole films. The polymer films grown in the ionic liquids show higher conductivity and better mechanical behaviour than those prepared in conventional solvents.68
The synthesis of poly(3-(4-fluorophenyl)thiophene) in the ionic liquids 1-ethyl-2,3-dimethylimidazolium bis(trifluoromethylsulfonyl) imide and 1,3-diethyl-5-methylimidazolium bis(trifluoromethylsulfonyl) imide was reported.69 Also, there are some recent studies on the synthesis of poly(3-(4-fluorophenyl)thiophene) in ionic liquids.70–73 MacFarlane and co-workers reported the electropolymerization of thiophene, bithiophene and terthiophene using the ionic liquids 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide and 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl) imide as the growth medium and supporting electrolyte.74 They reported also the synthesis of poly(3,4-ethylenedioxy) thiophene in the same ionic liquids.75 Quite recently we reported the synthesis and characterization of poly(para)phenylene in the ionic liquid 1-hexyl-3-methylimidazolium tris(pentafluoroethyl) trifluorophosphate.76,77
4. Synthesis of colloidal nanoparticles
There are some studies available in the literature on the synthesis of stable crystalline nanoparticles in ionic liquids which are now emerging as an important class of catalysts for various reactions. Metal nanoparticles have unique electronic properties, chemical reactivity and potential applications due to the quantum size effect which is derived from a dramatic reduction of the number of free electrons in nanoparticles smaller than 5 nm.It was reported78 that very fine and stable nanoparticles of Ir(0) and Ru(0) with 2.0–2.5 nm diameters can be synthesized in the dry ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate by chemical reduction. The presence of water causes the partial decomposition of the ionic liquid with the formation of phosphates, HF and metal fluorides. The isolated nanoparticles can be redispersed in the ionic liquid, in acetone or used in solventless conditions for the liquid–liquid biphasic, homogeneous or heterogeneous hydrogenation of arenes under mild reaction conditions (75 °C and 4 atm).78 Moreover, these catalytic systems can be recovered and reused several times.
Stable, isolable Pt(0) nanoparticles of 2–3 nm diameter and with a narrow size distribution can be easily obtained via decomposition of Pt-organometallic precoursors, e.g., Pt2(dba)3 (dba = bis-dibenzylidene acetone), in 1-butyl-3-methylimidazolium hexafluorophosphate ionic liquid.79 These nanoparticles are recyclable catalytic systems for the solventless or biphasic hydrogenation of alkenes and arenes under mild reaction conditions. The catalytic activity of the Pt nanoparticles is higher than that obtained for the classical PtO2 catalyst under the same reaction conditions.79
Itoh et al.80 reported the synthesis and functionalization of gold nanoparticles modified with ionic liquids based on the imidazolium cation. The obtained gold nanoparticles can be used as exceptionally high extinction dyes for colourimetric sensing of anions in water via particle aggregation process.80 Gold and platinum nanoparticles with diameters of 2–3.5 and 2–3.2 nm, respectively, can also be synthesized using novel thiol-functionalized ionic liquids (TFILs).81 TFILs act as a highly effective medium for the preparation and stablization of gold and platinum nanoparticles, thus becoming highly dispersible in aqueous media.81
Zhou and Antonietti reported on a low temperature synthesis of crystalline TiO2 nanoparticles in ionic liquids.82 TiO2 nanoparticles of 2–3 nm diameter and with surface areas of 554 m2 g−1 were obtained by stoichiometric hydrolysis of titanium tetrachloride in 1-butyl-3-methylimidazolium tetrafluoroborate (water-poor conditions) at 80 °C.82 This material is expected to have potential in solar energy conversion, catalysis, and optoelectronic devices. The simplicity of the preparation method reflects the advantage of the use of ionic liquids since they facilitate direct synthesis of crystalline species under ambient conditions.
Quite recently it was demonstrated that nanorods, hyperbranched nanorods and nanoparticles with different CoPt compositions can be synthesized in 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide.83 To get more information on the synthesis of functional nanoparticles and other inorganic nanostructures we would like to refer to the minireview of Antonietti.84
5. Carbon nanotubes
Since the discovery of carbon nanotubes (CNT) in 1991 by S. Iijima85 they have attracted considerable attention due to their unique properties. Carbon nanotubes are long graphitic thin cylinders which, if simplified, can be regarded as a sheet of graphite rolled into a cylinder. They can have a single cylindrical wall (SWNTs) or multiple walls (MWNTs), cylinders inside the other cylinders. Carbon nanotubes are currently being studied in an effort to understand their novel structural, electronic, and mechanical properties and to explore their huge potential for many applications in nanoelectronics,86 and as actuators87 and sensors.88 Electrodes composed of carbon nanotubes have generated much interest because of their high conductivity, large surface area89,90 and their ability to facilitate catalytic processes.91Recently, carbon nanotubes and ionic liquids have generated great interest in many areas. It was found that the use of ionic liquids as electrolytes for electrochemical applications involving carbon nanotube electrodes has proved possible and advantageous.92 The good electrochemical behaviour of carbon nanotube electrodes in ionic liquids, coupled to the wider electrochemical window and nonvolatility of these electrolytes, suggests new approaches for the design of capacitors, batteries and electromechanical actuators.92
It was shown that some ionic liquids exhibit unexpected strong interactions with carbon nanotubes, forming ionic gels after grinding together.93,94 Usui et al.95 prepared ionic nanocomposite gel electrolytes by dispersing carbon nanotubes into the ionic liquid [EMIm] Tf2N and assembled dye-sensitized solar cells (DSCs) using these electrolytes. They found that the energy conversion efficiency of DSCs prepared using such ionic electrolytes improved.95 Wallace et al. reported the mechanical properties of carbon nanotube electrodes in the ionic liquid [BMIm] BF4.96 They found that the ionic liquid interacts strongly with the carbon nanotubes affecting the mechanical properties of the electrodes. It was also reported that carbon nanotubes can be doped by ionic liquids.97
6. Batteries
Lithium batteries are used widely in portable electronic devices and electric vehicles. They show the highest energy density among the applicable chemical and electrochemical energy storage systems (up to 180 Wh kg−1). It is necessary that solvents for the electrolytes in Li-batteries are aprotic because of the requirements of wide electrochemical windows up to the cathodic limit of Li/Li+ potential. As known, the aprotic organic solvents are usually volatile and flammable. Therefore, the use of ionic liquids as electrolytes in Li-batteries is very promising. Matsumoto et al.98,99 applied several kinds of ionic liquids consisting of quaternary ammonium cation and imide anions to the classical lithium cell and they found that the ionic liquid 1-propyl-1-methylpiperidinium bis(trifluoromethylsulfonyl) imide is the most promising candidate as the electrolyte base. Nakagawa et al.100 reported that the use of the binary electrolyte [EMIm]BF4–LiBF4 shows high thermal stability and better electrochemical performance. Batteries using the ionic liquid [EMIm]Tf2N containing LiTf2N show better performance and low self-discharge.101 The self-discharge of the cell after 2000 h is less than 5% per month, which means that little corrosion and degradation of cell components take place.101 MacFarlane and co-workers102 investigated the ionic liquid [BMP]Tf2N containing LiTf2N for use as an electrolyte in Li-batteries. It was reported that the ionic liquid [EMIm] Tf2N shows a good electrolyte performance in Li–air batteries.103 A new ionic mixture composed of LiTf2N and acetamide was prepared and characterized as an electrolyte for Li batteries, too.104 The LiTf2N/acetamide mixture is liquid at room temperature between the molar ratios of 1 ∶ 2 and 1 ∶ 6, and it can be suggested for potential applications as lithium battery electrolytes.1047. Spectroscopy
Many papers were published on the spectroscopy of ionic liquids using several spectroscopic techniques, such as infrared (IR), ultraviolet (UV), optical Kerr effect (OKE), ultraviolet photoemission spectroscopy (UPS), mass spectroscopy (MS), Raman, fluorescence, in order to study the molecular and electronic structures, molecular dynamics and possible interactions. Some available results from the literature will be presented in this section.7.1. IR and Raman spectroscopy
There are some IR and Raman spectroscopy studies in ionic liquids. In these studies, information was provided in order to understand at molecular level the general interactions that exist in ionic liquids.Talaty et al.105 measured IR and Raman spectra of a series of 1-alkyl-3-methylimidazolium hexafluorophosphate ([C2−4Mim]PF6 ionic liquids and correlated the results with those obtained from calculations. These ionic liquids have common Raman C–H stretching frequencies that may serve as possible probes in studies of ionic liquid interactions. Hydrogen bonding interactions were observed between the fluorine atoms of the PF−6 anion and the C2 hydrogen on the imidazolium ring, and between PF−6 anion and the H atoms on the adjacent alkyl side chains.105
In situ Fourier transform infrared reflection absorption spectroscopy (FT-IRAS) was utilized to study the molecular structure of the electrified interphase between the ionic liquid [EMIm]BF4 and gold substrate.106 The feature in the FT-IRA spectra suggested that [EMIm]+ is adsorbed at the interphase and orients vertically with the molecular axis in the imidazolium ring nearly parallel to the electrode surface in a potential range of −1.3 V to +0.6 V vs. Ag/Ag+.106
Tran et al.107 employed near-infrared spectroscopy (NIR) technique for the noninvasive and in situ determination of concentrations and structure of water absorbed by the ionic liquids [BMIm]BF4, [BMIm]PF6 and [BMIm]Tf2N. It was found that absorbed water interacts with the anions of the ionic liquids; [BF4]− provides the strongest interactions and [PF6]− the weakest. In 24 h, [BMIm]BF4 can absorb up to 0.320 M of water, whereas [BMIm]PF6 only absorbs 8.3 × 10−2 M of water107 at the same time. Furthermore, they demonstrated that it is possible to use the NIR technique not only to characterize aggregation of surfactants in ionic liquids but also to determine kinetics and to identify products of reactions in ionic liquids as well as in microreactors provided by micelles in ionic liquids.108 NIR spectroscopy technique was used for sensitive and direct determination of critical micelle concentration (cmc) values of various nonionic surfactants in the ionic liquids [BMIm]PF6 and [EMIm]Tf2N.108
Raman investigation of the ionic liquid 1-propyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl) imide ([PMP]Tf2N) and its 2/1 mixture with LiTf2N was reported.109 The results showed that the [Tf2N]− anions have only a very weak interaction with the [PMP]+ cations, sterically shielded, but strong coordination to the Li+ cations.109
7.2. UPS, UV and fluorescence spectroscopy
Ultraviolet photoemission spectroscopy (UPS) is one of the powerful methods to probe the electronic structures of materials. However, it is usually difficult to apply to liquid samples due to their high vapour pressure. The usually extremely low vapour pressure of ionic liquids gives rise to applying the UPS technique to study electronic structures of ionic liquids, even under ultra-high vacuum conditions. Yoshimura et al.110 studied the electronic structures of the ionic liquids [BMIm]BF4, [BMIm]PF6 and [BMIm]Tf2N by UPS with synchrotron radiation.110 They found that the top of the valence states in the liquids is derived from the organic cation, although the highest occupied molecular orbitals (HOMOs) of the isolated anions are higher than that of the isolated cation.110The optical properties of [BMIm]PF6, [BMIm]BF4 and [EMIm]BF4 were recently investigated.111,112 The results showed that all imidazolium-based ionic liquids have significant absorption in the entire UV region and a long absorption tail that extends into the visible region. Furthermore, they all exhibit a very interesting excitation wavelength dependent fluorescence behaviour. Billard et al.113 demonstrated the importance of the purity of the ionic liquid [BMIm]PF6 in spectroscopic studies and showed that purification procedures suppress the absorption in the range 250–300 nm and beyond.
Fluorescence spectroscopy is a very useful technique to investigate molecular dynamics, molecular association, and microstructure within organized media. Recently, fluorescence techniques have been employed to characterize physicochemical properties of ionic liquids. Using fluorescence technique, Pandey and coworkers114–116 reported that the physicochemical properties of [BMIm]PF6 are altered by the addition of cosolvents.
Alvaro et al.117 investigated the energy, hydrogen, and electron transfer reactions within [BMIm]PF6. They observed slow molecular diffusion and low oxygen solubility within this relatively high viscosity IL, as well as an increase in the lifetime of radical ions and the triplet excited state. During the investigation of the possibility for cellulase catalyzed reactions in ionic liquids, Rogers et al.118 studied enzyme stability within 1-butyl-3-methylimidazolium chloride using a fluorescence techniques.
7.3. OKE spectroscopy
There are a few studies on the use of optical heterodyne-detected Raman-induced Kerr effect spectroscopy (OHD-RIKES) to probe experimentally the intermolecular and orientation dynamics of ionic liquids. Quitevis and co-workers119 reported a study of the effect of the alkyl chain length on the low frequency (0–250 cm−1) spectra for a homologous series of the ionic liquids 1-alkyl-3-methylimidazolium bis(trifluoromethylsufonyl)imide, [CnMim]Tf2N, n = 2, 4, 5, 6, 8, 10. The study of the temperature dependence of the low-frequency spectrum of [C5MIm]Tf2N was also reported.120Using OHD-RIKES, Giraud et al.121 investigated the ultrafast solvent dynamics of some ionic liquids, [BMMIm]Tf2N, [BMIm]PF6, [BMIm]Tf2N, [BMIm]TfO and [OMIm]Tf2N, by studying the effects of cation and anion substitution on the low-frequency spectra. It was found in all five samples that the signal is due to vibration of the imidazolium ring at three frequencies around 30, 65, and 100 cm−1 corresponding to three local configurations of the anion with respect to the cation.
7.4. Mass spectroscopy
Electrospray ionization mass spectroscopy (ESI-MS) was used to detect both the cations and anions of the ionic liquids as well as their solubility in water.122,123 It was found that in addition to the main peaks of the parent ions, fragmentation products are observed upon increasing the cone voltage, whereas aggregates of the parent ions with one or more ionic liquid molecules are observed upon decreasing the cone voltage. The main fragmentations of most studied ionic liquids were due to the loss of butene molecule.123Dyson et al.124 reported a dilution method for analyzing ionic liquids and catalysts dissolved in ionic liquids by ESI-MS. Jackson and Duckworth125 showed that the ionic liquids could be analysed without dilution using ESI-MS. They also demonstrated that ionic impurities or dissolved additives, especially those that are solvent reactive, could be detected overcoming the limitations of the dilution method.
Laser desorption/ionization (LDI) and matrix-assisted laser desorption/ionization (MALDI) mass spectroscopy are both methods which allow the investigation and characterization of ionic liquids. Tholey and co-workers126 used (LDI) and (MALDI) mass spectroscopy to characterize five different ionic liquids as well as studying the analysis of amino acids, peptides and proteins dissolved in these ionic liquids. Li and Gross127 tested some ionic liquids as MALDI matrices for quantification of peptides and proteins. Armstrong et al.128 introduced a class of specially designed ionic liquids that are capable of absorbing laser light and transferring protons to the analyte as matrices for MALDI mass spectroscopy.
8. Thermodynamics
Up to now, a number of papers have been published on the thermodynamic properties of ionic liquids. In this section we present some available literature data on the thermodynamic properties of ionic liquids.Thermodynamic activity coefficients are a measure of the deviation from ideal behaviour in liquid mixtures. Activity coefficients at infinite dilution can be directly used for the selection of solvents for extractive distillation, liquid extraction, solvent-aided crystallization, and even chemical reaction. Activity coefficients at infinite dilution give a direct measure of interactions between unlike molecules in the absence of solute–solute interactions.
Gas chromatography is widely used for determining thermodynamic properties of pure substances or solvent properties of binary mixtures. Using inverse gas chromatography, Mutelet and Jaubert129 determined the activity coefficients for 29 polar and non-polar compounds (alkanes, alkenes, alkynes, cycloalkanes, aromatics, alcohols) in the two following ionic liquids: 1-butyl-3-methylimidazolium octyl sulfate ([BMIm]+ [C8H17OSO3]−) at 323.15, 333.15, 343.15 K, and 1-ethyl-3-methylimidazolium tosylate ([EMIm]+[C7H7SO3]−) at 323.15 K.
Heintz and co-workers130 determined the activity coefficients at infinite dilution γ∞i of the linear and branched C1 to C6 alcohols, acetone, acetonitrile, ethylacetate, alkylethers, and chloromethane in the ionic liquid 4-methyl-N-butyl-pyridinium tetrafluoroborate by gas chromatography using the ionic liquid as the stationary phase. The partial molar excess enthalpies at infinite dilution HE,∞i of the polar solutes in the ionic liquid can be derived from the temperature dependence of the limiting activity coefficients. According to the Gibbs–Helmholtz equation, the value of HE,∞i can be directly obtained from the slope of a straight line derived from the following equation:

Vapour–liquid equilibria (VLE) of binary mixtures containing ethanol, propanol and benzene in the ionic liquids [BMIm]Tf2N131 and [EMIm]Tf2N132 were studied and the activity coefficients of these solvents in the ionic liquids were determined from VLE data.
The suitability of a solvent for separating mixtures of two components is defined as selectivity and can be determined using the equation:

The S∞12 values obtained for different binary mixtures indicated that the ionic liquid [EMIm]tosylate can play an important role for separation of aromatics, chloroalkanes and alcohols from alkanes.129 To get more information on the thermodynamics of non-aqueous mixtures containing ionic liquids we refer to a recently published review article.133
9. Catalysis
Nowadays, ionic liquids are widely used in catalysis not only as solvents or reaction media but also as catalysts, or catalyst activators. As there are a number of excellent reviews134–137 on the application of ionic liquids in catalysis and biocatalysis, we give here only a few examples of the use of some air and water stable ionic liquids in catalysis.MacFarlane et al.138 reported that dicyanamide based ionic liquids, [BMIm][dca] and [EMIm][dca], act as active base catalysts in the acetylation of alcohols. A suspension of palladium nanoparticles can be formed by reducing a solution of palladium acetate in the ionic liquid [BMIm]PF6 with H2. This recyclable catalytic system was used for the hydrogenation of alkenes,139 see section 4.
Welton and co-workers140 demonstrated the possibility of using a thermally controlled ionic liquid N-octyl-3-methylimidazolium tetrafluoroborate [C8C1Im]BF4)–water biphasic or homogeneous system for hydrogenation of but-2-yne-1,4-diol. The ionic liquid [C8C1Im]BF4 is immiscible with water at room temperature, but fully miscible at the reaction temperature of 80 °C. When the system is cooled to room temperature, it separates into two phases and the product is removed with the water phase and the catalyst remains in the ionic liquid. Favre et al.141 reported that a wide range of ionic liquids based on imidazolium and pyrrolidinium cations and weakly coordinating anions (such as BF−4, PF−6, Tf2N−, TfO−) proved to be efficient solvents for the biphasic rhodium catalyzed hydroformylation of 1-hexene.
Several aromatic aldehydes were oxidised in the ionic liquid [BMIm]PF6 using the catalyst Ni(acac)2 (acac = acetylacetonate) as the oxidant.142 The catalyst and ionic liquid could be recycled after extraction of the carboxylic acid product. The same catalytic system, ionic liquid and oxidant, was also used for the oxidation of ethylbenzene forming ethylbenzene hydroperoxide.143 Namboodiri et al.144 reported the oxidation of styrene to acetophenone ,the Wacker oxidation, in the ionic liquids [BMIm]BF4 and [BMIm]PF6 using PdCl2 as a catalyst. Seddon and Stark145 reported the oxidation of benzyl alcohol to benzaldehyde in imidazolium based ionic liquids by oxygen and a palladium acetate catalyst source.
Conclusion
In this review article we have tried to give an overview on the importance of ionic liquids in physical chemistry and we summarized literature until the end of 2005. Whereas ionic liquids were regarded as relatively new until about 2000 the situation has changed dramatically throughout the recent 3 years. In 2005 there were about 1500 peer reviewed papers on ionic liquids. The still rising interest in ionic liquids in various fields of chemistry will surely lead to a rising output of papers, stimulating further studies. It can be expected that ionic liquids develop to a main stream in various fields of chemistry and physical chemistry in the near future. We ourselves are very curious to see the future developments in this field and we are looking forward to many more papers dealing with these fascinating liquids.List of some abbreviations
| Abbreviation | Name |
|---|---|
| [BEIm] | 1-Butyl-3-ethylimidazolium |
| [BMIm]BF4 | 1-Butyl-3-methylimidazolium tetrafluoroborate |
| [BMIm]PF6 | 1-Butyl-3-methylimidazolium hexafluorophoshate |
| [BMIm]Tf2N | 1-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide |
| [BMP]Tf2N | 1-Butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl) imide |
| [BMMIm]Tf2N | 1-Butyl-2,3-dimethylimidazolium bis(trifluoromethylsulfonyl) imide |
| [BMIm]TfO | 1-Butyl-3-methylimidazolium trifluoromethanesulfonate |
| [C5MIm]Tf2N | 1-Pentyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide |
| [EEIm] | 1,3-Diethylimidazolium |
| [EMIm]BF4 | 1-Ethyl-3-methylimidazolium tetrafluoroborate |
| [EMIm]Cl | 1-Ethyl-3-methylimidazolium chloride |
| [EMIm][dca] | 1-Ethyl-3-methylimidazolium dicyanamide |
| [EMIm] PF6 | 1-Ethyl-3-methylimidazolium hexafluorophosphate |
| [OMIm]Tf2N | 1-Octyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide |
| [PMP] | 1-Propyl-1-methylpyrrolidinium |
| TFA | Trifluoroacetate |
| Tf2N | Bis(trifluoromethylsulfonyl) imide |
| TfO | Trifluoromethanesulfonate |
References
- P. Walden, Bull. Acad. Imper. Sci., 1914, 1, 1800 Search PubMed.
- F. H. Hurley and T. P. Weir, J. Electrochem. Soc., 1951, 98, 207 CrossRef CAS.
- H. L. Chum, V. R. Koch, L. L. Miller and R. A. Osteryoung, J. Am. Chem. Soc., 1975, 97, 3264 CrossRef CAS.
- J. Robinson and R. A. Osteryoung, J. Am. Chem. Soc., 1979, 101, 323 CrossRef CAS.
- J. S. Wilkes, J. A. Levisky, R. A. Wilson and C. L. Hussey, Inorg. Chem., 1982, 21, 1263 CrossRef CAS.
- T. B. Scheffler, C. L. Hussey, K. R. Seddon, C. M. Kear and P. D. Armitage, Inorg. Chem., 1983, 22, 2099 CrossRef CAS.
- D. Appleby, C. L. Hussey, K. R. Seddon and J. E. Turp, Nature, 1986, 323, 614 CrossRef CAS.
- C. L. Hussey, Adv. Molten Salt Chem., 1983, 5, 185 Search PubMed.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965 RSC.
- P. Bonhôte, A. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS.
- J. Fuller and R. T. Carlin, in Molten Salts, ed. P. C. Trulove, H. C. De Long, G. R. Stafford and S. Deki, PV 98-11, The Electrochemical Society Proceedings Series, Pennington, NJ, 1998, p. 227 Search PubMed.
- D. R. MacFarlane, P. Meakin, J. Sun, N. Amini and M. Forsyth, J. Phys. Chem. B, 1999, 103, 4164 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- “Ionic Liquids in Synthesis”, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2003 Search PubMed.
- Y. Ohno, Electrochemical Aspects Ionic Liquids, John Wiley & Sons, Inc., New Jersey, 2005 Search PubMed.
- P. C. Trulove and R. A. Mantz, in Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2003, pp. 103–126 Search PubMed.
- S. Carda-Broch, A. Berthod and D. W. Armstrong, Anal. Bioanal. Chem., 2003, 375, 191 CAS.
- K. R. Seddon, A. Stark and M. J. Torres, Pure Appl. Chem., 2000, 72, 2275 CrossRef CAS.
- P. A. Z. Suarez, S. Einloft, J. E. L. Dullius, R. F. De Souza and J. Dupont, J. Chim. Phys., 1998, 95, 1626 CAS.
- P. Wasserscheid, R. Van Hal and A. Boesmann, Green Chem., 2002, 4, 400 RSC.
- K. N. Marsh, J. A. Boxall and R. Lichtenthaler, Fluid Phase Equilib., 2004, 219, 93 CrossRef CAS.
- R. A. Mantz and P. C. Trulove, in Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2003, pp. 56–68 Search PubMed.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156 RSC.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef.
- J. Fuller, R. T. Carlin and R. A. Osteryoung, J. Electrochem. Soc., 1997, 144, 3881 CAS.
- J. D. Hlbrey and R. D. Rogers, in Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2003, pp. 41–55 Search PubMed.
- M. E. Van Valkenburg, R. L. Vaughn, M. Williams and J. S. Wilkes, 15th Symposium on Thermophysical Properties, 2003 Search PubMed.
- U. Shröder, J. D. Wadhawan, R. G. Compton, F. Marken, P. A. Z. Suarez, C. S. Consorti, R. F. de Souza and J. Dupont, New J. Chem., 2000, 24, 1009 RSC.
- Y. Katayama, S. Dan, T. Miura and T. Kishi, J. Electrochem. Soc., 2001, 148, C102 CrossRef CAS.
- P. He, H. T. Liu, Z. Y. Li, Y. Liu, X. D. Xu and J. H. Li, Langmuir, 2004, 20, 10260 CrossRef CAS.
- P.-Y. Chen and I.-W. Sun, Electrochim. Acta, 2000, 45, 3163 CrossRef CAS.
- P.-Y. Chen and I.-W. Sun, Electrochim. Acta, 1999, 45, 441 CrossRef CAS.
- M.-H. Yang and I.-W. Sun, J. Appl. Electrochem., 2003, 33, 1077 CrossRef CAS.
- M.-H. Yang, M.-C. Yang and I.-W. Sun, J. Electrochem. Soc., 2003, 150, C544 CrossRef CAS.
- S.-I. Hsiu and I.-W. Sun, J. Appl. Electrochem., 2004, 34, 1057 CrossRef CAS.
- I. Mukhopadhyay, C. L. Aravinda, D. Borissov and W. Freyland, Electrochim. Acta, 2005, 50, 1275 CrossRef CAS.
- N. Koura, T. Endo and Y. Idemoto, J. Non-Cryst. Solids, 1996, 205, 650 CrossRef.
- L. Simanavicius, A. Stakenas and A. Starkis, Electrochim. Acta, 1997, 42, 1581 CrossRef.
- Y.-F. Lin and I.-W. Sun, Electrochim. Acta, 1999, 44, 2771 CrossRef CAS.
- P.-Y. Chen, M.-C. Lin and I.-W. Sun, J. Electrochem. Soc., 2000, 147, 3350 CAS.
- P.-Y. Chen and I.-W. Sun, Electrochim. Acta, 2001, 46, 1169 CrossRef.
- S. I. Hsiu, J. F. Huang, I.-W. Sun, C. H. Yuan and J. Shiea, Electrochim. Acta, 2002, 47, 4367 CrossRef CAS.
- J. F. Huang and I.-W. Sun, Adv. Funct. Mater., 2005, 15, 989 CrossRef CAS.
- J. F. Huang and I.-W. Sun, Chem. Mater., 2004, 16, 1829 CrossRef CAS.
- H. Y. Hsu and C. C. Yang, Z. Naturforsch., B, 2003, 58b, 1055.
- J. F. Huang and I.-W. Sun, Eectrochim. Acta, 2004, 49, 3251 Search PubMed.
- J. F. Huang and I.-W. Sun, J. Electrochem. Soc., 2004, 151, C8 CrossRef CAS.
- J. F. Huang and I.-W. Sun, J. Electrochem. Soc., 2003, 150, E299 CrossRef CAS.
- J. F. Huang and I.-W. Sun, J. Electrochem. Soc., 2002, 149, E348 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, H. L. Munro, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2001, 7, 1010 RSC.
- A. P. Abbott, G. Capper, D. L. Davies and R. K. Rasheed, Chem.-Eur. J., 2004, 10, 3769 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed, J. Archer and C. John, Trans. Inst. Met. Finish., 2004, 82, 14 Search PubMed.
- S. Takeoka, M. Fujii, S. Hayashi and K. Yamamoto, Phys. Rev. B, 1998, 58, 7921 CrossRef CAS.
- F. Endres and S. Zein El Abedin, Phys. Chem. Chem. Phys., 2002, 4, 1649 RSC.
- F. Endres and S. Zein El Abedin, Phys. Chem. Chem. Phys., 2002, 4, 1640 RSC.
- F. Endres and S. Zein El Abedin, Chem. Commun., 2002, 8, 892 RSC.
- A. K. Agrawal and A. E. Austin, J. Electrochem. Soc., 1981, 128, 2292 CAS.
- J. Gobet and H. Tannenberger, J. Electrochem. Soc., 1986, 133, C322.
- J. Gobet and H. Tannenberger, J. Electrochem. Soc., 1988, 135, 109 CAS.
- T. Matsuda, S. Nakamura, K. Ide, K. Nyudo, S. J. Yae and Y. Nakato, Chem. Lett., 1996, 7, 569 CrossRef.
- Y. Katayama, M. Yokomizo, T. Miura and T. Kishi, Electrochemistry, 2001, 69, 834 Search PubMed.
- S. Zein El Abedin, N. Boressinko and F. Endres, Electrochem. Commun., 2004, 6, 510 CrossRef CAS.
- W. Freyland, C. A. Zell, S. Zein El Abedin and F. Endres, Electrochim. Acta, 2003, 48, 3053 CrossRef CAS.
- S. Zein El Abedin, H. K. Farag, E. M. Moustafa, U. Welz-Biermann and F. Endres, Phys. Chem. Chem. Phys., 2005, 7, 2333 RSC.
- Handbook of Conducting Polymers, ed. T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Marcel Dekker Inc., New York, 2nd edn., 1998 Search PubMed.
- K. Sekiguchi, M. Atobe and T. Fuchigami, J. Electroanal. Chem., 2003, 557, 1 CrossRef CAS.
- K. Sekiguchi, M. Atobe and T. Fuchigami, Electrochem. Commun., 2002, 4, 881 CrossRef CAS.
- J. M. Pringle, J. Efthimiadis, P. C. Howlett, J. Efthimiadis, D. R. MacFarlane and A. B. Chaplin et al., Polymer, 2004, 45, 1447 CrossRef CAS.
- E. Naudin, H. A. Ho, S. Branchaud, L. Breau and D. Belanger, J. Phys. Chem. B, 2002, 106, 10585 CrossRef CAS.
- H. Randriamahazaka, C. Plesse, D. Teyssie and C. Chevrot, Electrochem. Commun., 2003, 5, 613 CrossRef CAS.
- H. Randriamahazaka, C. Plesse, D. Teyssie and C. Chevrot, Electrochem. Commun., 2004, 6, 299 CrossRef CAS.
- P. Damlin, C. Kvarnström and A. Ivaska, J. Electroanal. Chem., 2004, 570, 113 CrossRef CAS.
- P. Danielsson, J. Bobacka and A. Ivaska, J. Solid State Electrochem., 2004, 8, 809 CAS.
- J. M. Pringle, M. Forsyth, D. R. MacFarlane, K. Wagner, S. B. Hall and D. L. Officer, Polymer, 2005, 46, 2047 CrossRef CAS.
- K. Wagner, J. M. Pringle, S. B. Hall, M. Forsyth, D. R. MacFarlane and D. L. Officer, Synth. Met., 2005, 153, 257 CrossRef CAS.
- S. Zein El Abedin, N. Borissenko and F. Endres, Electrochem. Commun., 2004, 6, 422 CrossRef CAS.
- O. Schneider, A. Bund, A. Ispas, N. Borissenko, S. Zein El Abedin and F. Endres, J. Phys. Chem. B, 2005, 109, 7159 CrossRef CAS.
- G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner, S. R. Teixera and J. Dupont, Chem.- Eur. J., 2003, 9, 3263 CrossRef CAS.
- C. W. Scheeren, G. Machado, J. Dupont, P. F. P. Fichtner and S. R. Teixera, Inorg. Chem., 2003, 42, 4738 CrossRef CAS.
- H. Itoh, K. Naka and Y. Chujo, J. Am. Chem. Soc., 2004, 126, 3026 CrossRef CAS.
- K.-S. Kim, D. Demberelnyamba and H. Lee, Langmuir, 2004, 20, 556 CrossRef CAS.
- Y. Zhou and M. Antonietti, J. Am. Chem. Soc., 2003, 125, 14960 CrossRef CAS.
- Y. Wang and H. Yang, J. Am. Chem. Soc., 2005, 127, 5316 CrossRef CAS.
- M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed., 2004, 43, 4988 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- P. G. Collins, M. S. Arnold and P. Avouris, Science, 2001, 292, 706 CrossRef.
- R. H. Baughman, C. T. Cui, A. A. Zakhidov, Z. Iqbal, J. N. Batisci, G. M. Spinks, G. G. Wallace, A. Mazzddi, D. De Rossi, A. G. Rinzler, O. Jaschinski, S. Roth and M. Kertesz, Science, 1999, 284, 1340 CrossRef CAS.
- J. Kong, N. R. Franklin, C. W. Zhou, M. G. Chopline, S. Peng, K. J. Cho and H. J. Dai, Science, 2000, 287, 622 CrossRef CAS.
- A. Srivastava, O. N. Srivastava, S. Talapatra, R. Vajtai and P. M. Ajayan, Nat. Mater., 2004, 3, 610 CrossRef CAS.
- A. Anson, J. Jagiello, J. B. Parra, M. L. Sanjuan, A. M. Benito, W. K. Maser and M. T. Martinez, J. Phys. Chem. B, 2004, 108, 15820 CrossRef CAS.
- K. Gong, Y. Dong, S. Xiong, Y. Chen and L. Mao, Biosens. Bioelectron., 2004, 20, 253 CrossRef CAS.
- J. N. Barisci, G. G. Wallace, D. R. MacFarlane and R. H. Baughman, Electrochem. Commun., 2004, 6, 22 CrossRef CAS.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072 CrossRef CAS.
- F. Zhao, X. Wu, M. Wang, Y. Liu, L. Gao and S. Dong, Anal. Chem., 2004, 76, 4960 CrossRef CAS.
- H. Usui, H. Matsui, N. Tanabe and S. Yanagida, J. Photochem. Photobiol. A: Chem., 2004, 164, 97 CrossRef.
- P. G. Whitten, G. M. Spinks and G. G. Wallace, Carbon, 2005, 43, 1891 CrossRef CAS.
- L. Kavan and L. Dunsch, ChemPhysChem, 2003, 4, 944 CrossRef.
- H. Sakaebe and H. Matsumoto, Electrochem. Commun., 2003, 5, 594 CrossRef CAS.
- H. Sakaebe, H. Matsumoto and K. Tatsumi, J. Power Sources, 2005, 146, 693 CrossRef CAS.
- H. Nakagawa, S. Izuchi, K. Kuwana, T. Nukuda and Aihara, J. Electrochem. Soc., 2003, 150, A695 CrossRef CAS.
- B. Garcia, S. Lavallee, G. Perron, C. Michot and M. Armand, Electrochim. Acta, 2004, 49, 4583 CAS.
- P. C. Howlett, D. R. MacFarlane and A. F. Hollenkamp, Electrochem. Solid-State Lett., 2004, 7, A97 CrossRef CAS.
- T. Kuboki, T. Okuyama, T. Ohsaki and N. Takami, J. Power Sources, 2005, 146, 766 CrossRef CAS.
- Y. Hu, H. Li, X. Huang and L. Chen, Electrochem. Commun., 2004, 6, 28 CrossRef CAS.
- E. R. Talaty, S. Raja, V. J. Storhaug, A. Dölle and W. R. Carper, J. Phys. Chem. B, 2004, 108, 13177 CrossRef CAS.
- N. Nanbu, Y. Sasaki and F. Kitamura, Electrochem. Commun., 2003, 5, 383 CrossRef CAS.
- C. D. Tran, S. H. D. Lacerda and D. Oliveira, Appl. Spectrosc., 2003, 57, 152 CrossRef CAS.
- C. D. Tran and S. Yu, J. Colloid Interface Sci., 2005, 283, 613 CrossRef CAS.
- M. Castriota, T. Caruso, R. G. Agostino, E. Cazzanelli, W. A. Henderson and S. Passerini, J. Phys. Chem. A, 2005, 109, 92 CrossRef CAS.
- D. Yoshimura, T. Yokoyama, T. Nishi, H. Ishii, R. Ozawa, H. Hamaguchi and K. Seki, J. Elect. Spectrosc. Related Phenom., 2005, 144–147, 319 Search PubMed.
- A. Paul, P. K. Mandal and A. Samanta, J. Phys. Chem. B, 2005, 109, 9148 CrossRef CAS.
- A. Paul, P. K. Mandal and A. Samanta, Chem. Phys. Lett., 2005, 402, 375 CrossRef CAS.
- I. Billard, G. Moutiers, A. Labet, A. El Azzi, C. Gaillard, C. Mariet and K. Lutzenkirchen, Inorg. Chem., 2003, 42, 1726 CrossRef CAS.
- K. A. Fletcher and S. Pandey, Appl. Spectrosc., 2002, 56, 266 CAS.
- K. A. Fletcher and S. Pandey, J. Phys. Chem. B, 2003, 107, 13532 CrossRef CAS.
- K. A. Fletcher, S. N. Baker, G. A. Baker and S. Pandey, New J. Chem., 2003, 27, 1706 RSC.
- M. Alvaro, B. Ferrer, H. Garcia and M. Narayana, Chem. Phys. Lett., 2002, 362, 435 CrossRef CAS.
- M. B. Turner, S. K. Spear, J. G. Huddleston, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 443 RSC.
- B. R. Hyun, S. V. Dzyuba, R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. A, 2002, 106, 7579 CrossRef CAS.
- J. R. Rajian, S. Li, R. A. Bartsch and E. L. Quitevis, Chem. Phys. Lett., 2004, 393, 372 CrossRef.
- G. Giraud, C. M. Gordon, I. R. Dunkin and K. Wynne, J. Chem. Phys., 2003, 119, 464 CrossRef CAS.
- Z. B. Alfassi, R. E. Huie, B. L. Milman and P. Neta, Anal. Bioanal. Chem., 2003, 377, 159 CrossRef CAS.
- B. L. Milman and Z. B. Alfassi, Eur. J. Mass Spectrom., 2005, 11, 35 CrossRef CAS.
- P. J. Dyson, J. S. McIndoe and D. Zhao, Chem. Commun., 2003, 508 RSC.
- G. P. Jackson and D. C. Duckworth, Chem. Commun., 2004, 522 RSC.
- M. Z. Moghaddam, R. Krüger, E. Heinzle and A. Tholey, J. Mass Spectrom., 2004, 39, 1494 CrossRef CAS.
- Y. L. Li and M. L. Gross, J. Am. Soc. Mass Spectrom., 2004, 15, 1833 CrossRef CAS.
- D. W. Armstrong, L.-K. Zhang, L. He and M. L. Gross, Anal. Chem., 2001, 73, 3679 CrossRef CAS.
- F. Mutelet and J.-N. Jaubert, J. Chromatogr., A Search PubMed , (in press).
- A. Heintz, D. V. Kulikov and S. P. Verevkin, J. Chem. Thermodyn., 2002, 34, 1341 CrossRef CAS.
- S. P. Verevkin, J. Safarov, E. Bich, E. Hassel and A. Heintz, Fluid Phase Equilib., 2005, 236, 222 CrossRef CAS.
- S. P. Verevkin, T. V. Vasiltsova, E. Bich and A. Heintz, Fluid Phase Equilib., 2004, 218, 165 CrossRef CAS.
- A. Heintz, J. Chem. Thermodyn., 2005, 37, 525 CrossRef CAS.
- T. Welton, Coord. Chem. Rev., 2004, 148, 2459 CrossRef.
- Z. Yang and W. Pan, Enzyme Microb. Technol., 2005, 37, 19 CrossRef CAS.
- D. Zhao, M. Wu, Y. Kou and E. Min, Catal. Today, 2002, 74, 157 CrossRef CAS.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef.
- S. A. Forsyth, D. R. MacFarlane, R. J. Thompson and M. von Ltzstein, Chem. Commun., 2002, 714 RSC.
- J. Huang, T. Jiang, B. Han, H. Gao, Y. Chang, G. Zhao and W. Wu, Chem. Commun., 2003, 1654 RSC.
- P. J. Dyson, D. J. Ellis and T. Welton, Can. J. Chem., 2001, 79, 705 CrossRef CAS.
- F. Favre, H. Olivier-Bourbigou, D. Commereuc and L. Saussine, Chem. Commun., 2001, 1360 RSC.
- J. Howarth, Tetrahedron Lett., 2000, 41, 6627 CrossRef CAS.
- R. Alcántara, L. Canoira, P. Guilherme-Joao and P. Pérez-Mendo, Appl. Catal., A, 2001, 218, 269 CrossRef CAS.
- V. V. Namboodiri, R. S. Varma, E. Sahle-Demessie and U. R. Pillai, Green Chem., 2002, 4, 170 RSC.
- K. R. Seddon and A. Stark, Green Chem., 2002, 4, 119 RSC.
Footnote |
| † Permanent address: Electrochemistry and Corrosion Laboratory, National Research Centre, Dokki, Cairo, Egypt. |
| This journal is © the Owner Societies 2006 |