Pressure perturbation calorimetric studies of the solvation properties and the thermal unfolding of proteins in solution—experiments and theoretical interpretation
Lally
Mitra
a,
Nikolai
Smolin
a,
Revanur
Ravindra
a,
Catherine
Royer
b and
Roland
Winter
*a
aUniversity of Dortmund, Department of Chemistry, Physical Chemistry I - Biophysical Chemistry, Otto-Hahn Str. 6, D-44227, Dortmund, Germany
bCentre de Biochimie Structurale, 29, rue de Navacelles, F-34090, Montpellier, France
First published on 19th January 2006
Abstract
We used pressure perturbation calorimetry (PPC), a relatively new and efficient technique, to study the solvation and volumetric properties of amino acids and peptides as well as of proteins in their native and unfolded state. In PPC, the coefficient of thermal expansion of the partial volume of the protein is deduced from the heat consumed or produced after small isothermal pressure jumps, which strongly depends on the interaction of the protein with the solvent or cosolvent at the protein–solvent interface. Furthermore, the effects of various chaotropic and kosmotropic cosolvents on the volume and expansivity changes of proteins were measured over a wide concentration range with high precision. Depending on the type of cosolvent and its concentration, specific differences were found for the solvation properties and unfolding behaviour of the proteins, and the volume change upon unfolding may even change sign. To yield a molecular interpretation of the different terms contributing to the partial protein volume and its temperature dependence, and hence a better understanding of the PPC data, molecular dynamics computer simulations on SNase were also carried out and compared with the experimental data. The PPC studies introduced aim to obtain more insight into the basic thermodynamic properties of protein solvation and volume effects accompanying structural transformations of proteins in various cosolvents on one hand, as these form the basis for understanding their physiological functions and their use in drug designing and formulations, but also to initiate further valuable applications in studies of other biomolecular and chemical systems.
1. Introduction
Understanding the stability and thermodynamics of the folded state of proteins has fascinated protein chemists and biophysicists for many years and still remains one of the most challenging issues in the field. In general, the stability of proteins depends on temperature, pressure, its hydration capacity and on the solvent properties.1–24 In this regard, volumetric properties of proteins, such as its partial specific volume, and even more significantly, the temperature and pressure derivatives thereof, have proven to be sensitive measures of hydration effects.6,23 It is well-known that the cytoplasm of the cell is relatively crowded, and surface to surface gaps of neighbouring organelles and biopolymers are, on average, less than 5 nm containing a rather complex solvent mixture.25–27 In the context of such proximity, we can expect the structure and dynamics of the solvent to be largely determined by the properties of the biomolecular surfaces, and—vice versa—the properties of the biomolecular systems will depend drastically on their direct solvent environment.Proteins in their native, folded configurations contain both hydrophilic and hydrophobic surface groups. As an illustrating example, Fig. 1 shows the surface groups and the first hydration layer of native SNase. Vicinal water molecules close to charged residues are expected to be oriented to the surface charges, leading to a more or less layered structure.23,28 In fact, this layering, if substantial, should be reflected in the thermodynamic properties of the protein, such as the partial molar volume and its apparent coefficient of thermal expansion, α. How many water layers with properties different from bulk solvent may form at the protein surface, depends principally on two factors: the charge density and its spatial distribution and the strength of the thermal forces that tend to disrupt the induced organization. Increase of temperature is expected to lead to a disruption of the solvent layer, which, in turn, should be reflected in the temperature dependence of α.
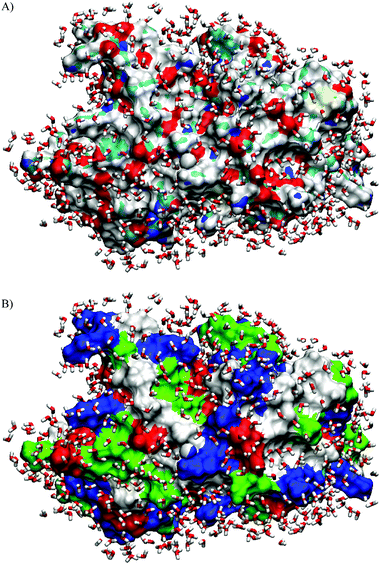 | ||
| Fig. 1 Arrangement of water molecules in the hydration shell from an MD simulation study on SNase (T = 300 K, cut-off distance from the protein surface: d = 4.5 Å). (A) The different types of atoms at the molecular surface are shown in different colours: N blue, O red, C cyan, S yellow and H white. (B) The various types of residues at the molecular surface of SNase are shown: nonpolar: white, acidic: red, basic: blue and polar: green). The hydration water molecules near the hydrophilic atoms are more tightly packed and exhibit a complex hydrogen bonding pattern, whereas the water molecules near the hydrophobic atoms are more loosely packed and form a hydrogen bonded network with chain-like arrangements of water molecules.36 | ||
The influence of the protein’s surface on adjacent water and how this might be reflected in the apparent expansivity data of proteins has been discussed extensively by Lin et al.29 Their seminal work indicates that exposed hydrophilic groups, such as charged or polar side chains of proteins, show a pattern characteristic of water structure breakers, with a large positive apparent expansion coefficient of the protein, in particular at low temperature, which decreases drastically with increasing temperature. On the contrary, apolar, hydrophobic amino acid side groups act in the reverse manner, as structure makers, by enhancing the space consuming hydrogen bonded network structure of water typical for hydrophobic hydration. Hence, for proteins in aqueous solution, the temperature dependent coefficient of thermal expansion is drastically influenced by protein–water interactions and therefore also by the magnitude and nature of the solvent accessible surface area (ASA) of the protein. Furthermore, irrespective of whether cosolvents directly bind to or are excluded from the protein surface, they are expected to induce significant changes in the quantity of “bound” water and its associated physical properties. In general, not only the degree of protein hydration, but also the structural and dynamic properties of the hydration layer change in the course of protein un- and refolding, aggregation, amyloidogenesis and binding events.
In this article, we review recent, published studies and introduce new work on PPC measurements of amino acids, tripeptides and proteins. The PPC technique is complementary to densimetric measurements23,30–34 and has the high sensitivity which is necessary to detect small volumetric changes. In fact, the relative volume changes of protein unfolding can be either positive or negative and are usually very small (ΔV/V < 1%). To complete the thermodynamic data, DSC results are given for some of the systems studied. In the first part, we introduce the PPC technique and discuss results on amino acids and tripeptides. We then present data on the solvation and volumetric properties of two enzymes, ribonuclease A (RNase A) and staphylococcal nuclease (SNase), in their native and unfolded states. Moreover, the effects of various chaotropic and kosmotropic cosolvents (glycerol, sorbitol, sucrose, urea, guanidinium hydrochloride (GuHCl), guanidinium sulfate (Gu2SO4), potassium sulfate (K2SO4), ethanol (EtOH), trifluoroethanol (TFE)) on the solvation and unfolding behaviour of the proteins is discussed. The influence of these cosolvents on α and hence on the hydrational properties of the protein provides insight into the underlying contributions to the volume changes accompanying unfolding. To yield a molecular interpretation of the different terms contributing to the partial protein volume and its temperature dependence and hence a better understanding of the PPC data, molecular dynamics (MD) computer simulations on SNase were also carried out and compared with experimental data.
2. Materials and methods
2.1 Materials
The amino acids alanine, leucine, glutamine, methionine and phenylalanine, of >99% minimum purity, were obtained from Sigma Aldrich, the tripeptides of >99% minimum purity from Bachem. All chemicals were dissolved in deionized water without further purification. The partial specific volumes of the amino acids and tripeptides were calculated according to the method of Reading et al.30 Bovine pancreatic ribonuclease A (RNase A) was purchased from Sigma Chemical Co. and used without further purification. Recombinant SNase with the sequence of nuclease A from the V8 strain of Staphylococcus aureus was obtained using the λ expression system in the Escherichia coli strain Arλ9 and purified as previously described.35,36 The salts, alcohols and osmolytes were obtained from Aldrich and used without further purification. Phosphate buffer (10 mM) (di-sodium hydrogen phosphate, Na2HPO4, anhydrous, from Merck) at pH 5.5 was used for all experiments, except where mentioned.2.2 DSC and PPC measurements
The differential scanning calorimetric traces were measured by means of a high precision VP DSC micro-calorimeter from MicroCal, Northampton, MA, USA. The reference cell was filled with matching buffer/cosolvent solution. Both buffer and protein solutions were degassed before being injected into the respective cells. The instrument is also equipped with a pressuring cap that allows application of 1.8 bar to the cells in order to avoid air bubbles at elevated temperatures. The instrument was operated in the high gain mode at a rate of 40 °C h−1. Baseline subtraction (pure buffer/cosolvent) and normalization with respect to the protein concentration were performed by the instrument software, yielding the temperature-dependent apparent molar heat capacity of the protein, Cp with respect to the buffer/cosolvent solution. The sample concentration of the proteins in 10 mM phosphate buffer was 0.5 wt% with a known quantity of cosolvent, except where mentioned.The pressure perturbation (PPC) experiments were performed in the DSC calorimeter using the MicroCal PPC accessory.11,29,37–40 The reference and sample cell volume are identical (0.5 mL) and they open to a common pressure chamber containing a sensor (Fig. 2A). The protein concentration used for the PPC studies was 5 mg mL−1, except where mentioned. An equal pressure of 5 bar was applied to both cells in a programmed manner using nitrogen gas (Fig. 2B). The software then initiates a pressure release to ambient pressure. The temperature is kept constant by active compensation of the heat change caused by the pressure jump. As the solutions in the sample and reference cell are identical except for the small amount of dissolved solute in the sample cell (solid ellipses in Fig. 2A) counterbalanced by the corresponding volume of buffer (at the same pH) in the reference cell (dashed ellipses), the measured differential heats are quite small. The compensation power returns to the baseline typically within 1 min and integration of the supplied power vs time yields the heat consumed or released by the sample. After complete equilibration, an opposite pressure jump is applied when the PPC controller reconnects the PPC cells with nitrogen gas. The heat peaks upon compression and decompression agree within a few % in absolute values, they are of opposite sign, however. For both compression and decompression experiments, temperature, pressure, and heat flow are recorded as a function of time. The calorimeter is then automatically heated or cooled to the next desired temperature and the next two (compression/decompression) pressure jumps are applied. Refs. 40–42 refer to an earlier debate concerning possible methodological errors within the PPC procedure.
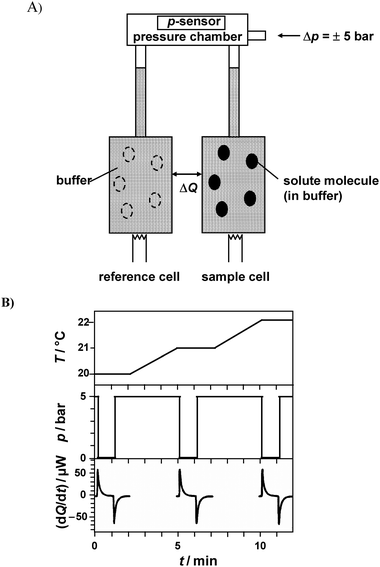 | ||
| Fig. 2 (A) Schematic drawing for the pressure perturbation calorimetry (PPC) experimental setup (adopted from refs. 37 and 38). The open ellipses in the reference cell represent the volume occupied by solvent in this cell, which counterbalances the volume occupied by the solute molecules in the sample (filled ellipses). (B) Time courses of the cell temperature T, pressure p, and the compensation power dQ/dt upon three (out of typically 20–80) pressure jumps (downward and upward). Before each pressure jump, equilibration of the calorimeter in the isothermal mode at the desired temperature (here 20, 21 and 22 °C) takes place. The compensation power returns to the baseline typically within 1 min and integration of the supplied power over time yields the heat consumed or released by the sample. The heat peaks caused by the upward and downward pressure jumps differ in sign but should agree in absolute values. Integration of dQ/dt yields two data points for Q(T) at a given pressure change. After equilibration, the calorimeter is automatically heated or cooled to the next desired temperature and the next compression/decompression cycle is repeated. | ||
Thermodynamics relates the pressure coefficient of the heat Qrev exchanged, (∂Qrev/∂p)T, in a reversible process to the coefficient of thermal expansion, α = (1/V)(∂V/∂T)p, of the sample volume. For a sufficiently dilute solution containing mS grams of solute and m0 grams of solvent, the volume is given by V = m0V0
+
ms![[V with combining macron]](https://www.rsc.org/images/entities/i_char_0056_0304.gif) s, where V0 is the specific volume of the solvent and s the partial specific volume of the solute. One then finds
s, where V0 is the specific volume of the solvent and s the partial specific volume of the solute. One then finds
(∂Qrev/∂p)T = −TVα = −T [m0V0α0
+
mss![[small alpha, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c2.gif) s ]. s ]. | (1) |
s = (1/s)(∂s/∂T)p are the coefficients of thermal expansion associated with the solvent volume and the solute partial volume, respectively.
In the actual differential PPC experiment, the volume occupied by the solute in the sample cell, mss, is replaced by the same volume of solvent in the reference cell. Within small pressure intervals (±5 bar here), the pressure dependence of V and α can be neglected for the systems studied here, and eqn (1) can be integrated to yield the working equation
| ΔQrev = −T [msss
−
mssα0 ] Δp | (2) |
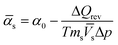 | (3) |
s, and the coefficient of thermal expansion, s, of the solute, will be replaced by their apparent values, denoted V and α, respectively. All reported PPC data are of the differential solution-versus-solvent type, adjusted to the same pH.
The relative volume changes ΔV/V at the unfolding transition of proteins, taking place in the temperature interval from To to Te, can be obtained by:
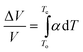 | (4) |
2.3 Molecular dynamics simulations of the protein–water interface
A series of MD simulations was performed on SNase in water at constant temperature and ambient pressure, namely at 300, 360 and 400 K. Details of the simulation method at ambient conditions can be found in our previous paper.43 Briefly, the MD simulations were performed using AMBER 6.0,44 the all-atom force field by Cornell et al.45 and the particle mesh Ewald (PME)46 was used for the calculation of electrostatic interactions. All protein atoms were explicitly included in the simulations and the TIP3P water model was used.47 All simulations were performed at constant N, p, T conditions and at a residue-based cut-off of 10 Å for van der Waals interactions. The simulations were continued for 7 to 13 ns. For the analysis, the trajectory from the last 2 ns was used. The program DSSP48 was used for the calculation of the solvent-accessible surface area (ASA). The van der Waals volume, VvdW, of the protein was calculated with the program Mol_Volume.49 The solvent-excluded volume, VSE, and the molecular surface area of the protein were determined using the program MSMS.50 A 1.4 Å radius for the probe sphere (H2O) was used for these calculations. To determine the standard errors of the mean values of fluctuating physical properties derived, we used the usual statistical analysis.51,523. Results and discussion
3.1 Amino acids
Fig. 3A displays selected curves of the temperature dependence of the thermal expansivity of alanine for varying concentrations. For all curves the global behaviour of expansivity of this amino acid in water is dominated by the structure breaking properties of the ionisable moieties. The value of α is large and positive at low temperature, reflecting the capacity of these ionisable moieties to cause a more dense organization of the hydrating water molecules around them compared to bulk water. The value of α decreases significantly as temperature increases, because the degree of interaction is diminished by increasing temperature.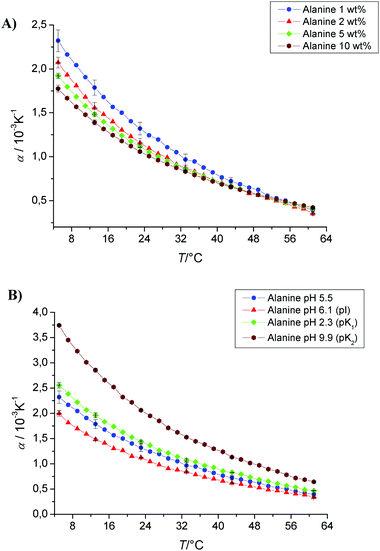 | ||
| Fig. 3 (A) Temperature dependence of the apparent thermal expansion coefficient α of alanine as a function of concentration (1, 2, 5 and 10 wt%) in phosphate buffer at pH 5.5. (B) Temperature dependence of the apparent thermal expansion coefficient α of alanine at different pH values, corresponding to its isoelectrical point (pI), pK1 and pK2 values (pI 6.1, pK1 2.3, pK2 9.9). | ||
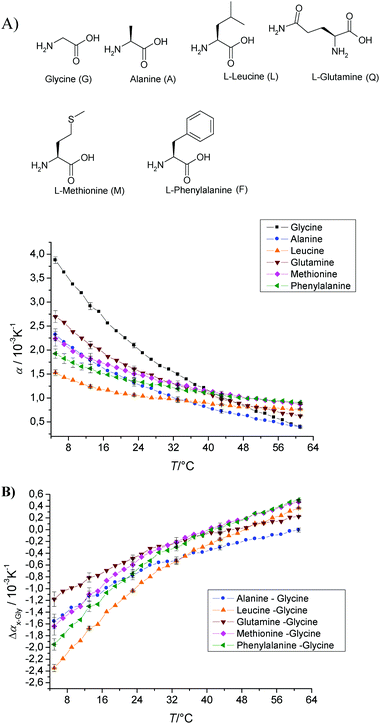 | ||
| Fig. 4 (A) Temperature dependence of the apparent thermal expansion coefficient α of various amino acids in 10 mM phosphate buffer at pH 5.5 and a concentration of 1 wt%. (B) To see the net side chain effect of the amino acids, the difference Δαx−Gly = αx − αGly is displayed. | ||
In order to eliminate the effect of the ionisable moieties on the value of α(T) and to evaluate the effect of the nature of the side chains R of the amino acids, the difference Δαx−Gly = αx − αGly was calculated (Fig. 4B). All glycine corrected curves start at negative Δαx−Gly values at low temperatures and reach positive values only at rather high temperatures >40 °C; the alanine side chain shows the most extreme behaviour, with Δαx−Gly becoming positive only at temperatures above 60 °C. Leucine exhibits the most negative value at low temperature, while that of glutamine is the smallest in absolute value.
It has been pointed out29 that solutes which increase the amount of structure in liquid water should have small positive or even negative α values at low temperature and a large positive temperature coefficient of α. Solutes that decrease the structure in water will show the opposite behaviour, with large positive α values at low temperature and a large negative temperature coefficient of α. This is simply the result of either increasing or decreasing, respectively, those features prominent in the behaviour of bulk liquid water that are attributable to tetrahedral structure melting.27,53 Such a scenario is fully consistent with our data on single amino acids, which show that Δα at low temperature becomes more negative with increased hydrophobicity of the a.a. side chain as measured by some hydrophobicity scale or transfer free energy measurement of a.a. side chains (e.g., from cyclohexane to H2O [kJ mol−1]:1 leucine, 20.59; phenylalanine, 12.47; methionine, 9.83; alanine, 7.57; glycine, 3.93; and glutamine: −23.18).
3.2 Tripeptides
Another means of evaluating the effect of the specific nature of the side chains of amino acids on their hydration and volumetric properties is to place the amino acid residue of interest in the context of a tripeptide, GXG, and subtract the PPC curve of the GGG tri-peptide from that of the GXG tri-peptide. This configuration has the advantage that the residue of interest is present in a configuration more closely resembling that of a polypeptide chain.31,32Fig. 5A shows the PPC curves of all tripeptides measured in sodium phosphate buffer at pH 5.5 and a concentration of 1 wt%. The coefficient of thermal expansion of GGG is the largest (α = 1.5 × 10−3 K−1) at low temperature, but still smaller than that of glycine (Fig. 3). All tripeptides show α values which are either smaller or larger than the corresponding single amino acids, except leucine where both α(T) values are similar. Fig. 5B shows the net effect of the side chains on the α values of the tripeptides (GXG) by subtracting the triglycine data (GGG): ΔαGXG−GGG = αGXG − αGGG (i.e., for comparing the α values of specific GXG tripeptides with the corresponding value of triglycine, we assumed—in a first approximation—additivity of α values). In comparison to the single amino acids, all tripeptides show less negative α values (e.g., the side chain of alanine has an α value of about −1.5 × 10−3 K−1, whereas α = −1.5 × 10−3 K−1 for the alanine side chain in GAG at T = 7 °C, i.e., there is a shielding effect of the neighbouring amino acids in the tripeptide, rendering the differential hydration contribution of the central a.a. residue to α less hydrophobic. Leucine shows the most negative α values for both the single amino acid side chain and for the tripeptide side chain with α values of −2.4 × 10−3 K−1 and −1.4 × 10−3 K−1, respectively, again indicating that the nearby hydration layers of the flanking amino acids markedly change (in this case decrease) the contribution of the central a.a. residue to α.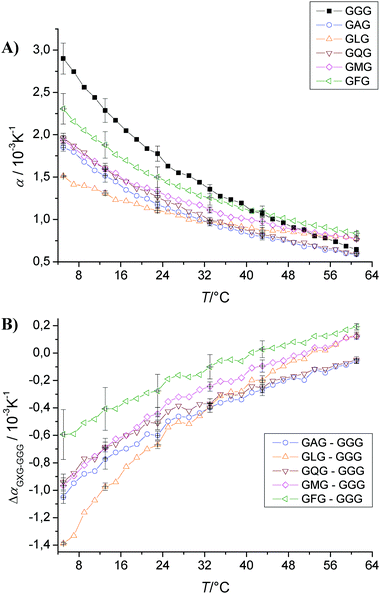 | ||
| Fig. 5 (A) Temperature dependence of the apparent thermal expansion coefficient α of substituted triglycine in 10 mM phosphate buffer at pH 5.5 and a concentration of 1 wt%. (B) To visualize the net effect of the side chains on the α values of the tripeptides (GXG), we show the subtracted triglycine data (GGG), ΔαGXG−GGG = αGXG − αGGG. | ||
The temperature dependence of the partial molar volumes of tripeptides of the sequence GXG has also been determined using differential scanning densimetry in order to evaluate a group additivity scheme for unfolded proteins.31,32 It is apparent from a perusal of the temperature dependence of the amino acid side-chain volumes in these studies that the side-chains may also be divided into two groups, typically hydrophobic (“structure-making” in terms of their effect on the solvent) and polar/ionic (“structure-breaking”), which have characteristic volume–temperature profiles with slopes which are qualitatively similar to our PPC data.31,32
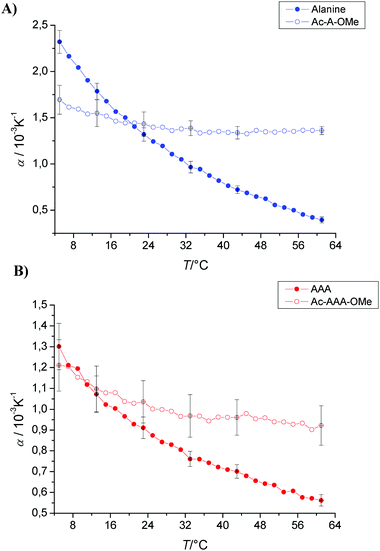 | ||
| Fig. 6 Temperature dependence of the apparent thermal expansion coefficient α of (A) alanine and (B) trialanine with “blocked” ends measured in phosphate buffer at pH 5.5 and a concentration of 1 wt%. | ||
3.3 Proteins
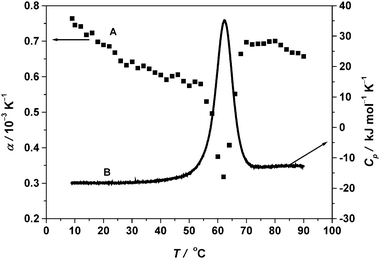 | ||
| Fig. 7 (A) PPC temperature dependence of the apparent thermal expansion coefficient α. (B) Background corrected DSC spectrum (scan rate 40 °C h−1) of 5 mg mL−1 RNase A in 10 mM phosphate buffer at pH 5.5. | ||
Using the post- and pre-transition base lines extrapolated to Tm, the change in α upon unfolding, Δα = 1.6 × 10−4 K−1 is obtained. The magnitude of Δα can be attributed to the exposure of most of the buried groups during thermal unfolding of RNase A. Lin et al.29 determined also α of reduced carboxymethylated (largely unfolded) RNase A between 4 and 95 °C from PPC measurements and found a value for Δα10−40 of approximately 3.0 × 10−4 K−1, which is much larger (43%) than that of native RNase A. From this value, the ASA of the native protein can be estimated as 43% of that accessible to the reduced unfolded form.
The area between the experimental data and the progress base line drawn between pre- and post-transition base lines projected into the transition region is utilized to calculate the area under the transitional peak (eqn (4)). The fractional volume change of unfolding, ΔV/V is obtained by integrating α over the temperature range where the transition occurs. By doing so, we obtain ΔV/V = −2.7 × 10−3 (−0.27%) for RNase A. The absolute volume change upon unfolding, ΔV, can be calculated from ΔV/V using the molar mass of RNase A (13.7 kDa) and its partial specific volume,54 which yields ΔV = −26 mL mol−1. Using densimetric measurements, at pH 2.5, also a negative volume change of unfolding has been observed for RNase A, however of smaller amplitude (ΔV = −10 mL mol−1).23 The observed negative volume change upon protein unfolding might be due to the opening of void volume and the electrostriction effect of the polar and charged groups of the increased ASA, overcompensating the positive effects of thermal volume and the changes in the hydrophilic–hydrophobic balance of the surface groups. The quality of the data thus clearly shows that, even with the low protein concentration chosen, the method is efficient enough to detect these small volume changes.
From the corresponding DSC data, we can derive the midpoint of the thermal unfolding temperature (Tm) and the area under the transition curve, the enthalpy change of the transition, ΔH. The enthalpy change obtained by integration over the DSC peak amounts to 435 ± 4 kJ mol−1. We can find an increase in Cp of 5.2 ± 0.2 kJ mol−1 K−1 between the unfolded and native state, a value which is typical for proteins and indicates an increased number of water–biopolymer contacts and hence hydration contribution during unfolding as also seen from the PPC graph. All these parameters derived from the PPC and DSC experiments for RNase A are taken as a reference to discuss their relative changes in the presence of various cosolvents (Table 1).
| Solvent | PPC Results | DSC Results | |||||||
|---|---|---|---|---|---|---|---|---|---|
| T m/ °C | α 10/ 10−3 K−1 | Δα10−40/ 10−4 K−1 | ΔV/V/ 10−3 | ΔV/ mL mol−1 | Δα/ 10−4 K−1 | T m/ °C | ΔH/ kJ mol−1 | ΔCp/ kJ K−1 mol−1 | |
| RNase A (5.5 mg mL−1) | 62.5 | 0.76 | 1.3 | −2.7 | −26.0 | 1.6 | 62.0 | 435 | 5.2 |
| RNase A (12.5 mg mL−1) | 63.0 | 0.66 | 0.7 | −2.6 | −25.0 | 1.5 | 62.1 | 424 | 5.4 |
| RNase A (21.0 mg mL−1) | 63.0 | 0.62 | 0.6 | −2.5 | −24.0 | 1.3 | 62.1 | 420 | 5.4 |
| + 0.5 M GuHCl | 57.0 | 0.5 | 0.3 | −3 | −29.0 | 2.1 | 57.8 | 349 | 3.1 |
| + 1.5 M GuHCl | 48.2 | 0.53 | 0.2 | −3.5 | −34.0 | 1.8 | 48.5 | 295 | 3.9 |
| + 2.5 M GuHCl | 40.0 | 0.74 | ∼0 | −3.6 | −35.0 | 0.8 | 40.1 | 226 | 4.6 |
| + 0.5 M Sucrose | 64.0 | 0.84 | 1.7 | −1.9 | −18.3 | 1.2 | 64.2 | 436 | 2.2 |
| + 1.5 M Sucrose | 68.1 | 0.68 | 1.3 | +1.3 | +12.5 | 0.8 | 67.8 | 448 | 1.6 |
| + 0.5 M Gu2SO4 | 61.0 | 0.6 | −0.2 | −2.2 | −21.2 | 1.3 | 61.5 | 371 | 2.6 |
| + 1.5 M Gu2SO4 | 61.5 | 0.58 | ∼0.4 | 0 | 0 | 0.9 | 62.0 | 385 | 3.3 |
| + 2.5 M Gu2SO4 | 65.0 | 0.54 | ∼0 | +1.5 | +14.5 | 1.0 | 66.0 | 376 | 3.5 |
| + 0.5 M EtOH | 60.5 | 0.84 | 2.1 | −2.6 | –25.1 | 1.6 | 60.6 | 405 | 4.0 |
| + 1.5 M EtOH | 59.5 | 0.84 | 1.6 | −2.2 | −21.2 | 1.5 | 58.6 | 420 | 1.0 |
| + 2.5 M EtOH | 57 | 0.86 | 1.4 | −2.0 | −19.3 | 1.4 | 56.8 | 456 | 3.4 |
| + 3.5 M EtOH | 55 | 0.92 | 0.9 | −1.7 | −16.4 | 1.2 | 54.8 | 440 | 4.0 |
| + 4.5 M EtOH | 53 | 1.3 | 2.9 | −1.4 | −13.5 | 1.0 | 54.0 | 457 | 4.4 |
| + 0.5 M TFE | 61.5 | 0.6 | 1.0 | −1.6 | +15.4 | 1.26 | 60.0 | 436 | 3.3 |
| + 1.5 M TFE | 55.1 | 0.46 | 0.9 | ∼0 | ∼0 | 0.93 | 56.0 | 453 | 3.4 |
| + 2.5 M TFE | 51.5 | −0.05 | 1.6 | +1.9 | +18.3 | 0.75 | 52.9 | 427 | 3.2 |
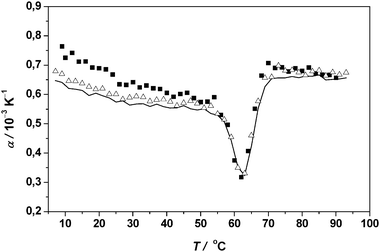 | ||
| Fig. 8 Temperature dependence of the apparent thermal expansion coefficient α of 5 (filled squares), 12.5 (open triangles) and 21 mg mL−1 RNase A (continuous line) in 10 mM phosphate buffer solution at pH 5.5. | ||
As an example, we show PPC experiments at 0.5 M (∼17 wt%) and 1.5 M (∼51 wt%) sucrose as cosolvent in 10 mM phosphate buffer. As can be clearly seen in Fig. 9, the hydration changes due to sucrose are significant. The comparison of α and (dα/dT)10–40 values of RNase A/0.5 M sucrose (−5.7 × 10−6 K−2) and RNase A in pure buffer solution (−4.3 × 10−6 K−2) indicates that there is a higher solvation (stronger hydration layer) of the native protein in the sucrose containing solvent, which also acts as an effective stabilizing agent as indicated by the higher Tm values (Fig. 9B, Table 1). The volume change upon unfolding is much smaller, −18.3 mL mol−1 in 0.5 M sucrose as compared to −26.0 mL mol−1 in the pure phosphate buffer system, which may indicate a less disordered unfolded state structure. This decrease in the absolute value of ΔV is also due to the fact that unfolding occurs at higher temperature and that the magnitude of ΔV decreases with increasing temperature. Above a sucrose concentration of ∼1 M, the volume change upon unfolding of the protein actually changes its sign; for example, in the 1.5 M sucrose solution, ΔV = +12.5 mL mol−1. Interestingly, at high sucrose concentrations, α decreases markedly, even below the value in the pure buffer system, indicating a significant decrease in protein hydration at these high cosolvent concentrations where the properties of bulk water are largely altered and specific cosolute binding to the protein becomes more likely. Also increased osmolyte-driven protein–protein interactions cannot be ruled out. According to the DSC data (Fig. 9B), the Tm value increases from 62 °C to 64 and 68 °C for the 0.5 and 1.5 M sucrose-RNase A systems, respectively. An incomplete thermal unfolding is also suggested by the reduced ΔCp values (Table 1).
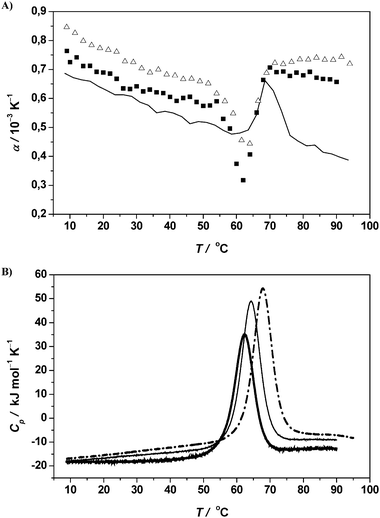 | ||
| Fig. 9 (A) Temperature dependence of the apparent thermal expansion coefficient α of RNase A (protein concentration 5 mg mL−1) in 10 mM phosphate buffer solution at pH 5.5 (filled squares) as well as in 0.5 M (open triangles) and 1.5 M (continuous thin line) sucrose in 10 mM phosphate buffer solution at pH 5.5. (B) Corresponding DSC traces of RNase A (thick line) and in the presence of 0.5 M (thin line) and 1.5 M (dashed dot line) sucrose. | ||
The PPC data of further osmolytes are discussed elsewhere 38 (see also Fig. 10). Briefly, for glycerol, the steric exclusion effect is expected to be smaller and as a result, hydration changes and stabilization due to glycerol are less pronounced. Sorbitol takes an intermediate position with regard to solvation of the native protein, to the protein stability, and the effect of cosolvent concentration on ΔV/V. With increasing cosolvent concentration, also for these polyols the volume change of unfolding becomes positive, however at much higher molar concentrations. A similar behaviour is observed for SNase.39
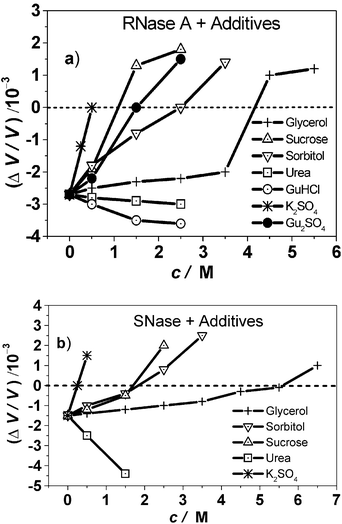 | ||
| Fig. 10 Relative volume changes, ΔV/V, of (a) RNase A and (b) SNase upon unfolding (protein concentration 5 mg mL−1, in 10 mM phosphate buffer solution) for various cosolvents as a function of cosolvent concentration. Please note that the unfolding temperatures Tm also vary as a function of cosolvent concentration (Table 1). | ||
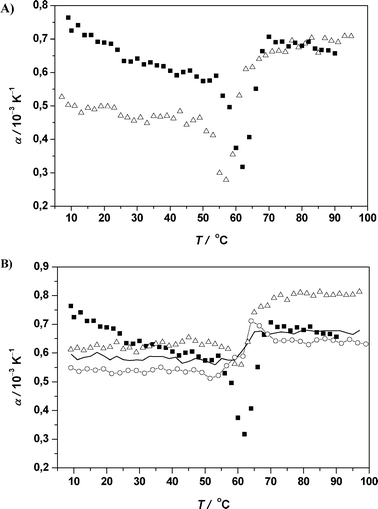 | ||
| Fig. 11 (A) Temperature dependence of the apparent thermal expansion coefficient α of RNase A (protein concentration 5 mg mL−1) in 10 mM phosphate buffer solution at pH 5.5 (filled squares) and in 0.5 M GuHCl/10 mM phosphate buffer solution at pH 5.5 (open triangles). (B) Temperature dependence of the apparent thermal expansion coefficient α of RNase A (protein concentration 5 mg mL−1) in 10 mM phosphate buffer solution at pH 5.5 (filled squares) and in 0.5 M (open triangles), 1.5 M (continuous thin line) and 2.5 M Gu2SO4 (open circles) in 10 mM phosphate buffer solution at pH 5.5. | ||
The lowered Tm value of 57.8 °C obtained from the DSC trace is in close agreement with the PPC result. ΔH of 349 kJ mol−1 in the presence of 0.5 M GuHCl is ∼20% smaller than the enthalpy change of unfolding of RNase A in pure buffer solution.
The major mechanism of stabilization by salt anions is known to be due to a reduction of the net positive charge of positively charged amino acid side groups on the protein surface through charge screening. Hofmeister anions,9,62,63 such as sulfate, are thought to also weakly interact with the peptide groups, whereas they interact unfavourably with nonpolar peptide side chains. From the PPC results for RNase A dissolved in 0.5 M K2SO4 buffer solution, we have shown38 that the shift in the Tm value from 62.0 to 69.0 °C is an indication that the salt acts as a strong stabilizing agent. By contrast to the osmolytes glycerol, sorbitol and sucrose, the apparent expansion coefficient of the native protein is markedly smaller, even smaller than that of the protein in pure buffer solution. Hence, even though the low α-value indicates a drastic net reduction in hydration, similar to urea and GuHCl, the K2SO4/buffer system increases the stability of the protein.
Here we show additional data where we replaced K+ by the strongly chaotropic guanidinium cation. We varied the Gu2SO4 concentration from 0.5 to 2.5 mol and the results of PPC and DSC scans are presented in Fig. 11B. It appears that at low concentrations, the guanidinium cations destabilize the protein as is visible by a slight shift of the Tm value to 64 °C. However, above 1.5 M Gu2SO4, the protein is stabilized and Tm increases to 66 °C at 2.5 M Gu2SO4, where the strongly stabilizing sulfate anions overcome the denaturing effect of the guanidinium cations. A decrease in the magnitude of ΔV and a reduction in ΔH values is observed (Table 1), both indicating that the protein is undergoing an incomplete unfolding. Above ∼0.5 mol Gu2SO4, the solvent contributions to α, and Δα10−40 are small. We should emphasize at this point, however, that a direct correlation between the denaturation volume of proteins and the denatured state structure is difficult and has to be treated with care owing to the different contributions to V and the complex interplay of these contributions. The water structuring tendencies are nearly eliminated in 2.8 M guanidinium sulfate where a long-range hydrogen-bonded water structure is largely absent. An almost constant value of α near 0.55 × 10−3 K−1 for native RNase A in 2.5 M Gu2SO4 buffer may represent the expansion of the intrinsic protein volume.
Thus, at high enough sulfate concentrations, the destabilizing effect of the guanidinium cation to native proteins is overcompensated by the strongly stabilizing and compaction-driving SO42− anion. As a consequence, also the ΔV value changes sign at high Gu2SO4 concentration, due, again, to a decrease in the magnitude of ΔV with increasing temperature and an increased compaction of the unfolded state. The negative contribution of the loss of void volume is minimized under these conditions, and therefore the positive contribution of the exposure of hydrophobic residues, coupled to the decrease in hydration of the polar moieties due to binding of the additive, may lead to the overall increase in volume upon unfolding, as has been observed before.29
Fig. 12A presents PPC scans of RNase A as a function of ethanol concentration in 10 mM phosphate buffer at pH 5.5 (for all the thermodynamic data derived from PPC and DSC, see Table 1). The reduced Tm values are an indication of the slight destabilising effect of ethanol. Interestingly, the magnitude of the reduction in volume is smaller than that of RNase A in pure buffer at a higher temperature, even at 4.5 M ethanol, indicating that the protein unfolds only partially. The solvent contribution to α and Δα10−40 is enhanced with increasing ethanol concentration, however. Hence, in contrast to the case of GuHCl, while like GuHCl ethanol displays a slightly destabilizing effect on Tm, the hydration contribution of the protein to the value and temperature dependence of α is increased.
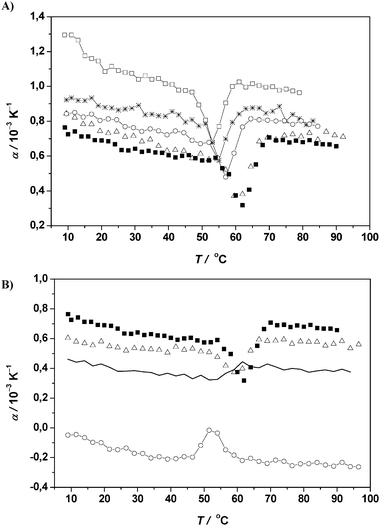 | ||
| Fig. 12 (A) Temperature dependence of the apparent thermal expansion coefficient α of RNase A (protein concentration 5 mg mL−1) in 10 mM phosphate buffer solution at pH 5.5 (filled squares) and in 0.5 M (open triangles), 2.5 M (open circles), 3.5 M (stars) and 4.5 M ethanol (open squares) in 10 mM phosphate buffer solution at pH 5.5. (B) Temperature dependence of the apparent thermal expansion coefficient α of RNase A (protein concentration 5 mg mL−1) in 10 mM phosphate buffer solution at pH 5.5 (filled squares) and in 0.5 M (open triangles), 1.5 M (continuous thin line), and 2.5 M TFE (open circles) in 10 mM phosphate buffer solution at pH 5.5. | ||
We have also studied the effect of 0.5 to 3.5 M TFE on RNase A solutions (Fig. 12B, Table 1). As can be clearly seen, TFE destabilizes the protein and, contrary to the cosolvent EtOH, it drastically reduces the hydration around the protein, as can be seen by a drastic reduction in α values. Surprisingly, at and above 1.5 M TFE, the volume change of unfolding, ΔV, of the protein changes sign and becomes positive. Hence, not only stabilizing osmolytes at high concentration, but also alcohols are able to induce positive ΔV values. The striking difference between the ethanol and TFE destabilisation scenarios is that TFE greatly disturbs the hydration layer around the protein by binding to the protein, thus drastically destabilising the protein’s tertiary structure. The ΔH and ΔCp values support the aforementioned conclusion that TFE greatly reduces the hydration by preferential solvation without marked unfolding of the secondary structure elements. A mere protein swelling (molten globule kind of state) upon denaturation might explain the positive ΔV values at high TFE concentrations.
3.4 Contributions to the partial protein volume and its coefficient of thermal expansion for native and unfolding proteins
As previously discussed by Chalikian,6 changes in partial specific volume, V, and hence in α of a protein can be considered to be dissected into essentially three different contributions: (1) the intrinsic volume, Vintr, which originates from the van der Waals volume of the constituent atoms plus the volume of intrinsic voids within the water-inaccessible protein interior; (2) a hydrational term, ΔVh, or “interaction volume”, describing—with regard to the bulk solvent—the solvent volume associated with the hydration of solvent-accessible protein atomic groups, resulting from solute–solvent interactions around the charged (electrostriction), polar (hydrogen-bonding), and nonpolar (hydrophobic hydration) atomic groups on the protein surface; and (3) the thermal volume, Vtherm, which results from thermally induced mutual molecular vibrations and re-orientations of the solute and the solvent: V ≈ Vintr + ΔVh + Vtherm. The thermal expansivity of the protein interior is generally expected to be small, which has yet to be verified (see below). The thermal volume increases with temperature leading to a positive contribution to α. The solvation effect may be expected to decrease with increasing temperature, as thermal activation leads to a continuous release of the “condensed” layered water from the protein surface (but see MD simulation results below). Once the water is released, it no longer contributes to the partial volume of the protein and hence to α. Such behaviour for α(T) of proteins has indeed been found in our experiments, pointing to the fact that the hydrational term plays a major role in determining the apparent thermal expansion coefficient of proteins. The negative volume change of unfolding observed for the two proteins at the transition temperature is probably due to the opening of void volume and the increase in hydrophilic hydration of more charged and polar groups. In the unfolded state, α increases due to a large increase of ASA and hence of the hydration contribution to α, in addition to the positive effects of thermal volume and the increased contribution of hydrophobic hydration in the unfolded state.We have seen that ΔV drastically depends on the type of cosolvent. However, the volume change that accompanies the unfolding transition of proteins is expected to depend also on the temperature at which the transition occurs. This is obvious given the fact that the expansivities of the folded states of proteins are different from those of the unfolded state and highly temperature dependent. Since the temperature dependences of the specific volume of the two states are different, the difference in volume between these two states cannot be constant with temperature. In fact, since the addition of osmolytes (glycerol, sorbitol, sucrose) stabilizes the folded state, such that the Tm increases with increasing glycerol concentration, and the relative volume change upon unfolding of RNase A, d(ΔV/V)/dT, decreases roughly at a rate of 5 × 10−4 K−1 (Fig. 10). Since the volume change of unfolding is expected to decrease with increasing temperature (as noted above), we may conclude that at least part of the observed effect of the stabilizing cosolvents on ΔV is due to the increase in transition temperature (leading to an increase of Vtherm), and not only due to differential conformational or hydrational effects. In fact, such an explanation should also hold for the observed slight increase in the absolute value of ΔV as a function of increasing urea or GuHCl concentration (d(ΔV/V)/dT ≈ 4.7 × 10−5 K−1 for RNase A, Fig. 10), as these cosolvents destabilize the folded state of the protein leading to a decrease in the Tm with increasing denaturant concentration.
Drastic temperature dependence of V would also be consistent with the pH dependence of the PPC curves of RNase A (data not shown). For example, the Tm value decreases from 63 °C at pH 7.7 (pI = 9.6) to 41 °C at pH 2, where the protein is strongly positively charged, and ΔV decreases concomitantly from −14.5 mL mol−1 at 63 °C to −38.6 mL mol−1 at 41 °C. The differences in d(ΔV/V)/dT values observed for the different cosolvents imply, however, that the different ΔV values observed for the various types of cosolvents can only partially be explained by the temperature dependence of ΔV. An additional contribution is probably due to a more or less completely unfolded state structure. Detailed small-angle scattering and CD/FT-IR spectroscopic measurements are needed to verify these assumptions.
3.5 Properties of the hydration water at the protein interface of SNase by MD computer simulations
To yield a molecular interpretation of the different terms contributing to the partial protein volume and its temperature dependence and hence a better understanding of the PPC data, MD computer simulations on SNase were also carried out and compared with the experimental data. Please note that a quantitative comparison of absolute volumetric and expansibility data with the experimental data is difficult owing to the fact that the absolute values of these parameters still depend on the water model and force field used in the MD simulation. In a recent MD study on SNase we studied in detail the water structure at the protein surface.43 We found that the structural properties of the water and hydrogen bonding pattern (network) at the protein surface are very complex and depend on its chemical and topographical properties. We classified the interfacial water molecules in the following way. A first type, water near hydrophobic residues, which induces bonding between neighbouring water molecules, gives raise to more pentagonal and hexagonal structural elements, which may grow into cage-like networks, forming disordered clathrate-like structures. A second type of interfacial water is the one near hydrophilic groups, such as charged or polar side chains of the protein. They interact strongly with water molecules, inducing a specific orientation of the water dipole. To investigate the influence of the protein surface on the structure of water, we calculated the local density profile of water as a function of the distance from the protein atoms. For precise evaluation of the water-density profile, we first calculated the volume of shells with thickness 0.1 Å as a function of the distance d from the nearest heavy atoms (N, O, C and S) of the protein. For calculation of the water local density profile, we counted the number of water molecules (the position of the water molecule was defined as the center of the oxygen atom) within the same shell and divided this number by the corresponding volume of their shell.43Fig. 13 shows the local density profiles of water from the simulation runs at ambient pressure and different temperatures, normalized by the density of bulk water at the same conditions. With increasing temperature, the height of the maximum of the water density profile, which corresponds to the hydration water (“bound water”), decreases and broadens, indicating a temperature-induced weakening of the water–protein interactions and smearing out of the structure of the hydration shell. Such behaviour should be reflected in the hydration contribution to α(T).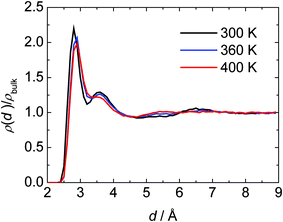 | ||
| Fig. 13 MD results for the local density profile of water, ρ(d), near the surface of SNase as a function of distance (d) from nearest protein heavy atoms, normalized by the density of bulk water at the same conditions. At p = 1 bar and T = 300, 360 and 400 K. | ||
In this study, we used one cut-off distance for all heavy atoms of the protein to define the hydration shell of SNase, and all atoms were used for calculating the physical surface properties of the protein molecule. Fig. 14 shows the average density of the hydration shell of SNase as a function of the cut-off distance d at different temperatures. A pronounced average density maximum close to the protein interface can be identified in all simulations, and appears at a similar distance of about 3.8 Å. As expected, this density maximum broadens with increasing temperature. About two water layers are perturbed at the protein surface and have different densities compared to bulk water.
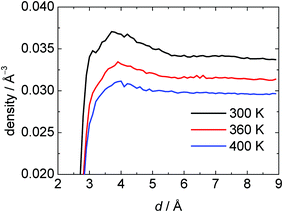 | ||
| Fig. 14 The average number density of the hydration water shell near the surface of SNase as a function of the cut-off distance (d) from nearest protein heavy atoms. At p = 1 bar and T = 300, 360 and 400 K. | ||
The thermal expansion coefficient, α = −ρ−1(∂ρ/∂T)p of the hydration shell has been determined from simulations at two (ρ, T) data points using α = −(2/(![[small rho, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d1.gif) 1
+
2)(2
−
1)/(T2
−
T1)) , where 1, 2 are the mean values of the density at temperatures T1 and T2, respectively. Fig. 15 exhibits the thermal expansion coefficient of the water within the hydration shell of SNase as a function of the cut-off distance, d. It can be clearly seen that α is much larger at low temperatures close to the protein interface, and drastically decreases at the higher temperature. To be able to compare the properties of the hydration and bulk water, we also determined α for the bulk TIP3P water by simulation of pure water under the same conditions (Table 2). The results are in agreement with previous simulations.78 TIP3P bulk water-like behaviour is observed above ∼5.5 Å
i.e., beyond the second hydration layer.
1
+
2)(2
−
1)/(T2
−
T1)) , where 1, 2 are the mean values of the density at temperatures T1 and T2, respectively. Fig. 15 exhibits the thermal expansion coefficient of the water within the hydration shell of SNase as a function of the cut-off distance, d. It can be clearly seen that α is much larger at low temperatures close to the protein interface, and drastically decreases at the higher temperature. To be able to compare the properties of the hydration and bulk water, we also determined α for the bulk TIP3P water by simulation of pure water under the same conditions (Table 2). The results are in agreement with previous simulations.78 TIP3P bulk water-like behaviour is observed above ∼5.5 Å
i.e., beyond the second hydration layer.
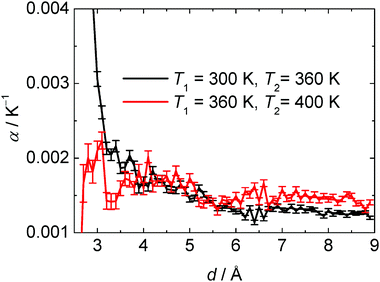 | ||
| Fig. 15 Thermal expansion coefficient α of the hydration shell of SNase as a function of the cut-off distance d from nearest protein heavy atoms. | ||
 | ||
| Fig. 16 Schematic representation of the solvent accessible, molecular, and van der Waals surfaces of a protein. | ||
The molecular and the solvent-accessible surface areas of SNase at different temperatures are shown in Table 3. In addition, we divided the ASA into different types of residues by their physicochemical properties: nonpolar, polar neutral, positively or negatively charged. The molecular and solvent-accessible surface areas of SNase slightly increase (∼0.8 and 0.3%, respectively) with increasing temperature to 360 K, but slightly decrease again at 400 K compared to ambient temperature conditions. The reason is a drastic surface area increase of polar neutral and nonpolar groups of about 8–10% and a concomitant, largely compensating decrease of surface areas of positively and negatively charged groups. Hence, a significant redistribution of surface residues is revealed, leading to a higher population of nonpolar and polar neutral residues. Upon further temperature increase, only a reduction in the number of nonpolar surface area essentially occurs. These findings probably depend on the specific surface topography of the protein under consideration.
| Area (Å2) | ||||||
|---|---|---|---|---|---|---|
| System | Molecular surface | Solvent-accessible surface | ||||
| All residues | Nonpolar | Polar, neutral | Positively charged | Negatively charged | ||
| SNase, 300 K, 1 bar | 7359 ± 12 (107) | 9417 ± 18 (160) | 1871 ± 7 (60) | 2271 ± 8 (70) | 3792 ± 8 (74) | 1483 ± 6 (50) |
| SNase, 360 K, 1 bar | 7418 ± 14 (131) | 9446 ± 24 (216) | 2054 ± 7 (66) | 2455 ± 11 (95) | 3615 ± 10 (90) | 1322 ± 9 (80) |
| SNase, 400 K, 1 bar | 7334 ± 14 (124) | 9395 ± 20 (180) | 1984 ± 8 (77) | 2468 ± 12 (110) | 3607 ± 11 (96) | 1336 ± 8 (72) |
For each type of protein surface group, we can define a corresponding protein volume, which is enclosed by the corresponding surface. The van der Waals volume, VvdW, of the protein is the volume enclosed within the van der Waals surface of the protein. The volume enclosed within the molecular surface is denoted solvent-excluded volume,75VSE, of the protein, which is equal to the sum of the van der Waals volumes of the protein and the volume of voids, Vvoid, of the protein resulting from imperfect internal packing. The partial volume, VP, of a protein in solution is defined as the volume of the solution minus the volume of the solvent in the absence of solute. The MD partial specific volume, VS = VP/mP, of SNase slightly changes from 0.680 cm3 g−1 at ambient temperature (T = 300 K) to 0.684 cm3 g−1 at T = 400 K (still far from MD unfolding conditions), respectively. Experimental data on SNase81 show a similar increase of VS(T) in the native state, though the absolute values are slightly larger (∼4%). At low temperatures, the temperature dependence of VS is larger than at higher temperatures just before the unfolding temperature. Such behaviour is in agreement with our simulations: with increasing temperature from 300 to 360 K, we obtain an increase of about 0.6% in the VS value. Further increase of temperature does not change the VS value significantly.
In Table 4, the various protein volumes of SNase obtained for the different temperatures are shown. In general, VP can be represented by the sum of the intrinsic geometrical volume, Vint = VvdW + Vvoid, occupied by the protein molecule itself (which can be approximated by the solvent excluded volume VSE of the protein molecule) and by the changes in the solvent volume, ΔVh, resulting from its interaction with accessible a. a. side chains, i.e., VP = VvdW + Vvoid + ΔVh.82,83 The hydration contribution, ΔVh, to the partial volume of the protein molecule reflects the interaction volume associated with the hydration of solvent accessible protein residues. ΔN = ΔVhρbulk is defined here as the number of water molecules that have to be added or released to the hydration shell in order to yield the bulk density value (ρbulk is the number density of water under the same temperature conditions).
| Volume/Å3 | ||||||
|---|---|---|---|---|---|---|
| System | Solvent-excluded volume, VSE | Van der Waals volume, VvdW | Void volume, Vvoid | Partial protein volume, Vp | Hydration contribution to the partial volume | ΔN = ΔVhρbulk |
| SNase, 300 K, 1 bar | 21482 ± 10 (86) | 18472 ± 2 (21) | 3010 ± 8 (73) | 18987 ± 43 (393) | −2495 ± 6 (52) | −82 |
| SNase, 360 K, 1 bar | 21604 ± 14 (109) | 18476 ± 2 (21) | 3127 ± 11 (100) | 19113 ± 49 (446) | −2491 ± 6 (58) | −77 |
| SNase, 400 K, 1 bar | 21665 ± 15 (111) | 18480 ± 2 (22) | 3186 ± 11 (99) | 19059 ± 56 (512) | −2606 ± 8 (70) | −76 |
Hence, at least for a semiquantitative interpretation of the partial volume of the protein in terms of hydration,84 we may use the following relationship
| VP = VSE + nh (Vh − V0), | (5) |
The partial volume in the simulation is simply the difference between the volumes of the system before and after adding the protein to the solution. We determined the partial volume of SNase from the simulations, using:
| VP = VBOX −mW/ρbulk − VCI, | (6) |
It is well known that for polar solutes, ΔVh is negative and for nonpolar solutes positive.86 Around a hydrophilic solute the water molecules are more tightly packed while in the presence of the hydrophobic solutes the water structure is less dense.86 For proteins, ΔVh is generally found to be negative. Remarkably, with increasing temperature, the absolute value of ΔVh increases (for nonpolar and polar solutes), as the decrease of the bulk water density is faster than the decrease of the hydration water density with increasing temperature. As expected, VvdW is more or less independent of temperature. Due to a marked increase of Vvoid (∼4%) up to 360 K, VSE of SNase increases (∼1%) with increasing temperature. The van der Waals and void volumes make up 86 and 14% of the solvent excluded volume, respectively, at T = 300 K. The void volume of the protein increases markedly with increasing temperature (∼4%), from 3010 Å3 at 300 K to 3127 Å3 at 360 K. For these two temperatures, the negative values of the hydration contribution to the partial volume are essentially the same, which is probably due to some surface rearrangement of the protein, leading to a decrease of the polar and concomitant increase of nonpolar and polar neutral protein surface area as discussed above (Table 3). This leads to a decreasing absolute value of ΔVh. At the higher temperature of 360 K, the magnitude of ΔVh increases in fact, as the decay of the bulk water density with increasing temperature is larger than the decay of the water density of the hydration water.
With further increasing the temperature from 360 to 400 K, a further but less pronounced increase of the void volume is visible. Obviously, the temperature dependence of the void volume is largest at lower temperatures and reaches a limiting value at high temperatures, probably since a further expansion without unfolding of the protein is not feasible any more. As the contribution of the polar and nonpolar amino acid residues at the protein surface remains the same at 360 and 400 K, we observe an increase of the absolute value of ΔVh due to the temperature increase in this case.
Additionally, we determined the thermal expansion coefficient of the different protein volumes (Table 5A) using the same dissection method as described above. In a first approximation, the thermal expansion coefficient of the protein partial volume may be expressed as:
| αVP = φVvdWαVvdW + φVvoidαVvoid + φΔVhαΔVh, | (7) |
| (a) α/10−3 K−1 | |||||
|---|---|---|---|---|---|
| SNase | Solvent excluded volume | Van der Waals volume | Void volume | Hydration contribution to the partial volume | Protein partial volume |
| T 1 = 300 K, T2 = 360 K | 0.094 ± 0.009 | 0.004 ± 0.003 | 0.63 ± 0.08 | ≈0 | 0.10 ± 0.01 |
| T 1 = 360 K, T2 = 400 K | 0.07 ± 0.02 | 0.005 ± 0.005 | 0.33 ± 0.13 | 1.13 ± 0.41 | −0.06 ± 0.03 |
| (b) φiαi/10−3 K−1 | |||||
|---|---|---|---|---|---|
| SNase | Van der Waals volume | Void volume | Hydration contribution to the partial volume | Protein partial volume | |
| T 1 = 300 K, T2 = 360 K | 0.00389 | 0.102 | ≈0 | 0.10 ± 0.01 | |
| T 1 = 360 K, T2 = 400 K | 0.00484 | 0.0546 | −0.151 | −0.06 ± 0.03 | |
As can be seen from Table 5B, for the temperature interval from 300 to 360 K, the main contribution to the expansion coefficient of the protein partial volume is due to the expansion of the void volume of the protein (0.13% K−1). At higher temperatures (360 to 400 K) we observe a competition between a decreasing but still positive contribution of the void volume (0.05% K−1) to the expansion coefficient of the protein and an increasing negative hydration contribution to the partial protein volume (−0.12% K−1). The drastic decrease of the expansion coefficient of the protein partial volume with increasing temperature, which has also been observed experimentally using PPC, can thus be attributed to a decrease of the expansibility of voids with increasing temperature and a decrease of the expansibility contribution of the hydration layer.
Finally, these volumetric data indicate that, because the void volume contribution of the unfolded state’s partial volume is probably negligible, the decrease in the absolute value of the volume change of unfolding, ΔV, with increasing temperature is essentially due to a decrease in the void volume and a more negative hydration contribution to the partial volume of the folded protein. The latter contributions also lead, as we have seen above (Table 5), to a decrease of the coefficient of thermal expansion of the folded protein at higher temperatures.
4. Concluding remarks
To conclude, pressure perturbation calorimetry has been found to be an effective tool to measure the apparent thermal expansion coefficient and volume changes of proteins in solution with very high precision, and valuable information related to volume changes upon unfolding and denaturation of proteins can be obtained. The thermodynamic data determined drastically depend on the interfacial solvent conditions, which influence the conformation as well as the stability of the biopolymer. The large body of results for various chaotropic and kosmotropic cosolvents clearly shows that the cosolvents change markedly not only the stability of a protein, but also its solvation, and hence may also alter the conformation of the protein in its unfolded, denatured state. The magnitude and even the sign of the volume change of unfolding drastically depend on the cosolvent concentration and strongly decrease with increasing temperature. The MD computer simulations revealed that the thermal expansion coefficient of the native protein’s partial volume is largely determined by the expansibility of its internal voids and by a significant hydration contribution, and both contributions decrease with increasing temperature. More generally, the present results demonstrate that the PPC technique provides an exquisitely sensitive handle on measurements of the hydration properties of biomolecules, the understanding of which is a prerequisite for the comprehension of their stability, structure and conformational dynamics. We may expect in the very near future that this method will show its great potential also in studies of proteins involved in more biologically and medically relevant research, such as in studies of amyloidogenesis of proteins.87–89Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (DFG) and the Fonds der Chemischen Industrie is gratefully acknowledged.References
- A. Fersht, Structure and Mechanism in Protein Science, W. H. Freeman and Co., New York, 1999 Search PubMed.
- S. N. Timasheff, Adv. Protein Chem., 1998, 51, 355–432 CAS.
- S. N. Timasheff, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 67–97 CrossRef CAS.
- S. N. Timasheff, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9721–9726 CrossRef CAS.
- G. Xie and S. N. Timasheff, Protein Sci., 1997, 6, 211–221 CAS.
- T. V. Chalikian, Annu. Rev. Biophys. Biomol. Struct., 2003, 32, 207–235 CrossRef CAS.
- S. K. Pal, J. Peon and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1763–1768 CrossRef CAS.
- V. A. Parsegian, R. P. Rand and D. C. Rau, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3987–3992 CrossRef CAS.
- M. G. Cacace, E. M. Landau and J. J. Ramsden, Q. Rev. Biophys., 1997, 20, 241–277 CrossRef CAS.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and F. G. Parak, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16047–16051 CrossRef CAS.
- R. Ravindra and R. Winter, ChemPhysChem, 2003, 4, 359–365 CrossRef CAS.
- W. Doster and M. Settles, Biochim. Biophys. Acta, 2005, 1749, 173–186 CAS.
- J. C. Smith, F. Merzel, A.-N. Bondar, A. Tournier and S. Fischer, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 1181–1190 CrossRef CAS.
- P. E. Smith, Biophys. Chem., 2005, 113, 299–302 CrossRef CAS.
- D. I. Svergun, S. Richards, M. H. J. Koch, Z. Sayers, S. Kuprin and G. Zaccai, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2267–2272 CrossRef CAS.
- R. B. Sessions, G. L. Thomas and M. J. Parker, J. Mol. Biol., 2004, 343, 1125–1133 CrossRef CAS.
- B. Halle, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 1207–1224 CrossRef CAS.
- S. Shimizu and D. J. Smith, J. Chem. Phys., 2004, 121, 1148–1154 CrossRef CAS.
- D. Russo, G. Hura and T. Head-Gordon, Biophys. J., 2004, 86, 1852–1862 CrossRef CAS.
- Advances in High Pressure Bioscience and Biotechnology II, ed. R. Winter, Springer-Verlag, Heidelberg, 2003 Search PubMed.
- S. Bergqvist, M. A. Williams, R. O’Brien and J. E. Ladbury, Mol. Biol., 2004, 336, 829–842 CrossRef CAS.
- A. Cooper, Biophys. Chem., 2005, 115, 89–97 CrossRef CAS.
- J. Rösgen and H.-J. Hinz, Biophys. Chem., 2000, 83, 61–71 CrossRef CAS.
- A. Oleinikova, I. Brovchenko, N. Smolin, A. Krukau, A. Geiger and R. Winter, Phys. Rev. Lett., 2005, 95, 247802 CrossRef.
- G. H. Pollack, Cells, Gels and the Engines of Life, Ebner & Sons, Seattle, WA, 2001 Search PubMed.
- P. Ball, Life’s Matrix: A Bibliography of Water, University of California Press, 2001 Search PubMed.
- F. Franks, Water: A Matrix of Life, The Royal Society of Chemistry, London, 2000 Search PubMed.
- M. F. Toney, J. N. Howard, J. Richer, G. L. Borges, J. G. Gordon, O. R. Melroy, D. G. Wiesler, D. Yee and L. B. Sorensen, Nature, 1994, 368, 444–446 CrossRef CAS.
- L.-N. Lin, J. F. Brandts, J. M. Brandts and V. Plotnikov, Anal. Biochem., 2002, 302, 144–160 CrossRef CAS.
- J. F. Reading and G. R. Hedwig, J. Chem. Soc., Faraday Trans., 1990, 86, 3117–3123 RSC.
- M. Häckel, H.-J. Hinz and G. R. Hedwig, Phys. Chem. Chem. Phys., 2000, 2, 4843–4849 RSC.
- M. Häckel, H.-J. Hinz and G. R. Hedwig, Biophys. Chem., 1999, 82, 35–50 CrossRef CAS.
- T. V. Chalikian, M. Totrov, T. Abagyan and K. J. Breslauer, J. Mol. Biol., 1996, 260, 588–603 CrossRef CAS.
- T. V. Chalikian and K. J. Breslauer, Biopolymers, 1996, 39, 619–626 CrossRef CAS.
- G. Panick, G. J. A. Vidugiris, R. Malessa, G. Rapp, R. Winter and C. A. Royer, Biochemistry, 1999, 38, 4157–4164 CrossRef CAS.
- G. Panick, R. Malessa, R. Winter, G. Rapp, K. J. Frye and C. A. Royer, J. Mol. Biol., 1998, 275, 389–402 CrossRef CAS.
- P. Kujawa and F. M. Winnik, Macromolecules, 2001, 34, 4130–4135 CrossRef CAS.
- R. Ravindra and R. Winter, Z. Phys. Chem., 2003, 217, 1221–1243 CrossRef CAS.
- R. Ravindra, C. Royer and R. Winter, Phys. Chem. Chem. Phys., 2004, 4, 1952–1961 RSC.
- H. Heerklotz, J. Phys.: Condens. Matter, 2004, 16, R441–R467 CrossRef CAS.
- S. L. Randzio, Thermochim. Acta, 2003, 398, 75–80 CrossRef CAS.
- J. F. Brandts and L.-N. Lin, Thermochim. Acta, 2004, 414, 95–100 CrossRef CAS.
- N. Smolin and R. Winter, J. Phys. Chem. B, 2004, 108, 15928–15937 CrossRef CAS.
- D. A. Case, D. A. Pearlman, J. W. Caldwell, T. E. Cheatham, III, W. S. Ross, C. L. Simmerling, T. A. Darden, K. M. Merz, R. V. Stanton, A. L. Cheng, J. J. Vincent, M. Crowley, V. Tsui, R. J. Radmer, Y. Duan, J. Pitera, I. Massova, G. L. Seibel, U. C. Singh, P. K. Weiner and P. A. Kollman, Amber, version 6, University of California, CA, San Francisco, 1999 Search PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS.
- Mol Volume—a program in the MDTools utility package at the Theoretical Biophysics group, a NIH Resource for Macromolecular Modeling and Bioinformatics, http://www.ks.uiuc.edu/Development/MDTools.
- M. F. Sanner, J.-C. Spehner and A. J. Olson, Biopolymers, 1996, 38, 305–320 CrossRef CAS.
- H. Flyvberg and H. G. Petersen, J. Chem. Phys., 1989, 91, 461–466 CrossRef CAS.
- D. Finchan, N. Quirke and D. J. Tildesley, J. Chem. Phys., 1986, 84, 4535–4546 CrossRef.
- R. Ludwig, Angew. Chem., Int. Ed., 2001, 40, 1808–1827 CrossRef CAS.
- K. Gekko and H. Noguchi, J. Phys. Chem., 1979, 83, 2706–2714 CrossRef CAS.
- K. Gekko and S. N. Timasheff, Biochemistry, 1981, 20, 4667–4676 CrossRef CAS.
- A. Priev, A. Almagor, S. Yedgar and B. Gavish, Biochemistry, 1996, 35, 2061–2066 CrossRef CAS.
- K. Gekko and T. Morikawa, J. Biochem., 1981, 90, 39–50 CAS.
- J. C. Lee and S. N. Timasheff, J. Biol. Chem., 1981, 256, 7193–7201 CAS.
- H. Betting, M. Häckel, H.-J. Hinz and M. Stockhausen, Phys. Chem. Chem. Phys., 2001, 3, 1688–1692 RSC.
- B. J. Bennion and V. Daggett, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5142–5147 CrossRef CAS.
- G. I. Makhatadze, J. Phys. Chem. B, 1999, 103, 4781–4785 CrossRef CAS.
- C. H. I. Ramos and R. L. Baldwin, Protein Sci., 2002, 11, 1771–1778 CrossRef CAS.
- K. D. Collins and W. Washabaugh, Q. Rev. Biophys., 1985, 18, 323–422 CrossRef CAS.
- A. C. Young, R. F. Tilton and J. C. Dewan, J. Mol. Biol., 1994, 235, 302–317 CAS.
- N. Hirota, K. Mizuno and Y. Goto, J. Mol. Biol., 1998, 275, 365–378 CrossRef CAS.
- M. Buck, Q. Rev. Biophys., 1998, 31, 297–355 CrossRef CAS.
- R. Walgers, T. C. Lee and A. Cammers-Goodwin, J. Am. Chem. Soc., 1998, 120, 5073–5079 CrossRef CAS.
- A. Kentsis and T. R. Sosnick, Biochemistry, 1998, 37, 14613–14622 CrossRef CAS.
- P. D. Thomas and K. A. Dill, Protein Sci., 1993, 2, 2050–2065 CrossRef CAS.
- M. Fioroni, M. D. Diaz, K. Burger and S. Berger, J. Am. Chem. Soc., 2002, 124, 7737–7744 CrossRef CAS.
- H. B. Bull and K. Breese, Biopolymers, 1978, 17, 2121–2131 CrossRef CAS.
- D.-P. Hong, M. Hoshino, R. Kuboi and Y. Goto, J. Am. Chem. Soc., 1999, 121, 8427–8433 CrossRef CAS.
- S. Cinelli, G. Onori and A. Santucci, J. Phys. Chem. B, 1997, 101, 8029–8034 CrossRef CAS.
- J. A. Rupley and G. Careri, Adv. Protein Chem., 1991, 41, 37–172 CAS.
- M. L. Connolly, J. Am. Chem. Soc., 1985, 107, 1118–1124 CrossRef CAS.
- F. Merzel and J. C. Smith, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5378–5383 CrossRef CAS.
- M. Levitt and R. Sharon, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 7557–7561 CAS.
- W. L. Jorgensen and C. Jenson, J. Comput. Chem., 1998, 19, 1179–1186 CrossRef CAS.
- B. K. Lee and F. M. Richards, J. Mol. Biol., 1971, 55, 379–380 CrossRef CAS.
- F. M. Richards, Annu. Rev. Biophys. Bioeng., 1977, 6, 151–176 Search PubMed.
- H. Seemann, R. Winter and C. A. Royer, J. Mol. Biol., 2001, 307, 1091–1102 CrossRef CAS.
- T. V. Chalikian and K. J. Breslauer, Biopolymers, 1996, 39, 619–626 CrossRef CAS.
- T. V. Chalikian and K. J. Breslauer, Curr. Opin. Struct. Biol., 1998, 8, 657–664 CrossRef CAS.
- F. J. Millero, A. Lo Surdo and C. Shin, J. Phys. Chem., 1978, 82, 784–792 CrossRef CAS.
- B. Lee, J. Phys. Chem., 1983, 87, 112–118 CrossRef CAS.
- V. Gutmann, Pure Appl. Chem., 1991, 63, 1715–1724 CrossRef CAS.
- Y. Cordeiro, J. Kraineva, R. Ravindra, L. M. T. R. Lima, M. P. B. Gomes, D. Foguel, R. Winter and J. L. Silva, J. Biol. Chem., 2004, 279, 32354–32359 CrossRef CAS.
- W. Dzwolak, R. Ravindra, C. Nicolini, R. Jansen and R. Winter, J. Am. Chem. Soc., 2004, 126, 3762–3768 CrossRef CAS.
- W. Dzwolak, S. Grudzielanek, V. Smirnovas, R. Ravindra, C. Nicolini, R. Jansen, A. Loksztejn, S. Porowski and R. Winter, Biochemistry, 2005, 44, 8948–8958 CrossRef CAS.
| This journal is © the Owner Societies 2006 |