Platinum dissolution and deposition in the polymer electrolyte membrane of a PEM fuel cell as studied by potential cycling
Kazuaki
Yasuda
*,
Akira
Taniguchi
,
Tomoki
Akita
,
Tsutomu
Ioroi
and
Zyun
Siroma
Research Institute for Ubiquitous Energy Devices, National Institute of Advanced Industrial Science and Technology (AIST), 1-8-31 Midorigaoka, Ikeda, Osaka, 563-8577, Japan
First published on 19th December 2005
Abstract
The behavior of platinum dissolution and deposition in the polymer electrolyte membrane of a membrane-electrode-assembly (MEA) for a proton-exchange membrane fuel cell (PEMFC) was studied using potential cycling experiment and high-resolution transmission electron microscopy (HRTEM). The electrochemically active surface area decreased depending on the cycle number and the upper potential limit. Platinum deposition was observed in the polymer electrolyte membrane near a cathode catalyst layer. Platinum deposition was accelerated by the presence of hydrogen transported through the membrane from an anode compartment. Platinum was transported across the membrane and deposited on the anode layer in the absence of hydrogen in the anode compartment. This deposition was also affected by the presence of oxygen in the cathode compartment.
Introduction
Recently, highly efficient and environmentally clean power generation has become a world-wide concern. Proton-exchange membrane fuel cells (PEMFCs), which electrochemically convert the chemical energy of a fuel directly into electrical energy, have gained attention as promising candidate power sources for electrically powered vehicles, small-scale stationary power generation and portable electronics devices in the near future.1–6 Reliability and working lifetime are the most important considerations in such power sources. However, there is as yet limited information available about failure modes in PEM fuel cells, and the causes and mechanisms of degradation are not fully understood.7 Scientific research and fundamental studies on phenomena that occur in an MEA are needed for the commercial success of PEMFC technology. Carbon black-supported platinum plays an important role as an electrocatalyst for both electrode reactions in PEMFC. Several studies have reported platinum sintering in the catalyst layer during PEMFC operation.8–10 In particular, the deterioration of the cathode kinetics by degradation of the cathode platinum is a major problem.Potential cycling is usually used for cleaning or activating an electrode surface before an electrochemical experiment. It is known that potential cycling of a platinum electrode to a higher potential region dissolves platinum.11–13 Ota et al. studied platinum dissolution in a sulfuric and phosphoric acid solution and reported that accelerated corrosion took place when oxides were formed and this could be reduced by cyclic potential sweeps.14 Patterson used this method to investigate the platinum dissolution in an MEA for use in PEMFC to accelerate the loss of surface area for a platinum electrocatalyst and to predict the effect of this loss of electrochemical area on PEMFC cell performance.15 Recently, Japan Storage Battery16 and UTC Fuel Cells17 have reported the durability evaluation of a PEMFC electrode using potential cycling.
Although platinum dissolution has mainly been studied in aqueous electrolyte solution and platinum sintering in the fuel cell electrode has been reported in PEMFC, the behavior of platinum that has dissolved and diffused into a polymer electrolyte membrane of PEMFC has not yet been studied. We reported that platinum aggregates are precipitated in the polymer electrolyte membrane during long-term operation of a PEMFC as part of a national PEMFC R&D project in Japan,18 but the mechanisms of these phenomena are not clear. We found that similar phenomena can be roughly reproduced by potential cycling. In this report, we have focused on the platinum dissolution from the cathode by potential cycling and studied the behavior of dissolved platinum in the polymer electrolyte membrane. We used high-resolution transmission electron microscopy (HRTEM), which is the most effective technique for investigating the distribution of nano-size platinum particles deposited in a polymer electrolyte membrane, since even a small amount of deposited platinum particle can be detected, although an analysis using an electron probe micro analyzer has also been reported. This work provides some key evidence regarding the behavior of dissolved platinum based on TEM characterization of damaged MEA samples created by diamond knife ultramicrotomy.
Experimental
Preparation of MEA
Electrodes for the PEMFC were prepared from a 40 wt% carbon black-supported platinum (Pt/C) electrocatalyst (Johnson Matthey) for the cathode, 30 wt% Pt/C/15 wt% Ru/C electrocatalyst (Johnson Matthey) for the anode and a Nafion® solution (5 weight percent solution, E.I. DuPont de Nemours and Company). Catalyst ink was prepared by adding Nafion® solution with isopropyl alcohol to the electrocatalyst powder. The resulting ink was applied on the polytetrafluoroethylene (PTFE) sheet followed by drying and transferred to polymer electrolyte membrane (Nafion® 117, 7 mil thick and 1100 EW sulfonic acid form, DuPont) by hot-pressing in order to manufacture a membrane electrode assembly (MEA). Nafion® content of electrodes were 15 wt%. Nafion® 112 (2 mil thick and 1100 EW sulfonic acid form, DuPont) was also used for comparison. Nafion® membrane was pretreated with hydrogen peroxide and 1 M H2SO4 aqueous solution. Wet-proofed carbon cloth (E-TEK ELAT) was used as gas diffusion backing.Potential cycling experiment
Experiments were carried out in a single circular cell (10 cm2) made from titanium without a precious metal coating (cell A as shown in Fig. 1) to avoid the effect of cell material corrosion. The flow field was a conventional parallel channel design.19,20 Potential cycling was carried out as follows. A two-electrode potentiostatic circuit was used; hydrated pure nitrogen was passed through the working electrode (fuel cell cathode) compartment and hydrated pure hydrogen went through the counter electrode (fuel cell anode) compartment. The cell was maintained at 80 °C and the operating pressure was atmospheric. Gases were humidified at the same temperature as the cell and fed to each electrode. The potential was cycled by a triangular wave at a scan rate of 50 mV s−1 using an electrochemical analyzer (BAS100B/W). The anode served as a reference electrode as well as a counter electrode. The electrochemically active surface area was evaluated from the hydrogen desorption charge of a cyclic voltammogram. | ||
| Fig. 1 Schematic drawing of single cell A and B used for the experiment. 1: proton exchange membrane (PEM), Nafion® 117, 2: anode (counter electrode) made of PtRu/C, 3: anode (counter electrode) made of carbon black without platinum, 4: cathode (working electrode) made of Pt/C, 5: titanium cell housing, 6: PTFE cell housing, 7: titanium current collector, 8: Viton® gasket, 9: liquid junction of 1 M H2SO4 aqueous solution, 10: reference electrode. | ||
For experiments without hydrogen, cell B connected to a standard hydrogen electrode by a H2SO4 electrolyte junction was used as shown in Fig. 1. The cell housing of cell B was made of PTFE and the current collectors were made of titanium. The periphery of the electrode was filled with 1 M H2SO4 aqueous solution. A Viton® gasket was located between the electrode and H2SO4 aqueous solution. The standard hydrogen electrode was connected to the membrane at the periphery of the electrode by 1 M H2SO4 aqueous solution, as shown in our previous papers.21,22Table 1 summarizes the potential cycling conditions of the analyzed samples discussed in this report.
| Sample name | Potential range/V vs. RHE | Cycle number | Scan rate/mV s−1 | Atmosphere cathode–anode | Membrane | Cell |
|---|---|---|---|---|---|---|
| a Cell B is equipped with a reference electrode and the anode (counter electrode) contains no platinum. | ||||||
| N-1 | 0.1–1.2 | 500 | 50 | N2–H2 | Nafion®117 | A |
| N-2 | 0.1–1.0 | 500 | 50 | N2–H2 | Nafion®117 | A |
| N-3 | 0.1–0.8 | 500 | 50 | N2–H2 | Nafion®117 | A |
| N-4 | 0.65–1.2 | 500 | 50 | N2–H2 | Nafion®117 | A |
| N-5 | 0.1–1.2 | 500 | 50 | N2–H2 | Nafion®112 | A |
| N-6 | 0.15–1.2 | 500 | 50 | N2–N2 | Nafion®117 | Ba |
| A-1 | 0.65–1.2 | 500 | 50 | Air–H2 | Nafion®117 | A |
Characterization by TEM, EDX and XPS
A small piece was removed from the MEA to prepare the TEM sample and carbon cloth was peeled from both planes of this piece. The cross-section of the MEA sample was prepared via epoxy impregnation and an ultramicrotome (Leica ULTRACUT UCT)-sectioning using a diamond knife. Samples for the TEM observations were directly supported on a ϕ 3 mm copper mesh with a carbon micro-grid. The thickness of the sample was about 30–60 nm. The TEM observation was performed using a JEOL JEM-3000F electron microscope at an accelerating voltage of 300 kV. The TEM was equipped with a nano-EDX (energy-dispersive X-ray spectroscopy) device for of elemental analysis in the nano-size region. Smearing of Pt particles into the MEA was not found in the TEM images of the samples before experiment and those after CV experiment in the potential scan range below 1.0 V vs. RHE. Therefore we think that Pt particles are not smeared into the MEA by the cutting process using ultramicrotome.Results and discussion
Platinum deposition in a polymer electrolyte membrane
Fig. 2 shows typical cyclic voltammograms of the cathode (working electrode) of the MEA as a result of the potential cycling from 0.1 V to 1.2 V vs. RHE in the cell A (sample N-1 in Table 1). In general, a cyclic voltammogram of a fuel cell electrode made from carbon black-supported platinum in the MEA does not show a clear characteristic voltammogram of platinum metal, as seen with a usual polycrystalline platinum metal electrode in an aqueous electrolyte solution. However, the cyclic voltammogram is useful for assessing the electrochemically active surface area of platinum in the MEA catalyst layer.23,24 This figure clearly shows that the electrochemically active surface area of the platinum catalyst decreases with an increase in the cycle number. | ||
| Fig. 2 Typical cyclic voltammograms for the potential cycling experiment ranging from 0.1 V to 1.2 V vs. RHE at 50 mV s−1 and 80 °C under a nitrogen atmosphere (sample N-1). | ||
The dependence of the electrochemically active surface area on the cycle number is shown in Fig. 3. The decrease in the electrochemically active surface area depended not only on the cycle number but also on the upper potential limit of the scanned potential region. The most significant loss was seen when the upper potential limit was 1.2 V vs. RHE (N-1). Qualitatively similar observations regarding a potential effect have been reported in studies on platinum dissolution in aqueous electrolyte solution.11,13 Two sintering mechanisms have been proposed to explain the loss of surface area of carbon-supported platinum in an aqueous electrolyte:25,26 dissolution/reprecipitation27–29 and migration (surface diffusion) of platinum particles.30,31 Although we can not exclude the possibility of platinum particle growth by a migration mechanism, we consider that the dissolution of platinum plays an important role in the decrease in the electrochemically active surface area. Some of the dissolved platinum ionic species such as Pt2+10–12 are redeposited on other platinum particles, resulting in platinum particle growth (electrochemical Ostwald ripening),26,29 and the remaining dissolved platinum ionic species diffuse out of the electrode and into the polymer electrolyte membrane.
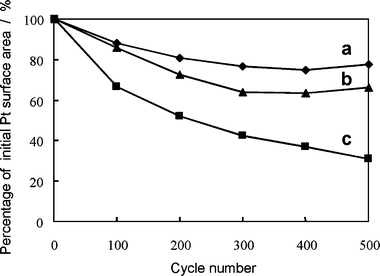 | ||
| Fig. 3 Dependence of the ratio of the electrochemically active surface area determined from cyclic voltammograms to the initial value on the cycle number (samples N-1, N-2 and N-3). Potential cycling range: (a) 0.1–0.8 V, (b) 0.1–1.0 V, (c) 0.1–1.2 V vs. RHE. Membrane: Nafion® 117. Atmosphere: nitrogen. | ||
Fig. 4a–c shows TEM images near the cathode catalyst layer of the cross-section of the MEA. A cathode catalyst layer and Nafion® membrane are seen on a net-like structure of a carbon micro-grid in these figures. The upper right side of the TEM image is a Pt/C catalyst layer and Pt particles are seen as dark contrast in the TEM image. The lower left side is a polymer electrolyte membrane region, which shows uniform contrast at this magnification. Platinum aggregates (aggregate-like particles) were deposited in the polymer electrolyte membrane near the cathode interface in samples with an upper potential limit of 1.2 V (N-1) and 1.0 V (N-2). The amount of platinum particles in the membrane increased with an increase in the upper potential limit from 1.0 V to 1.2 V. A high-resolution image of a platinum particle near the cathode interface of N-2 is shown in Fig. 5. Agglomerates seem to be comprised of particles of a few tens of nm in size. EDX measurements confirmed that these particles are composed of platinum. These platinum particles are much larger than the platinum catalyst particles loaded in the catalyst layer. On the other hand, potential cycling under an upper potential limit of 0.8 V (N-3) resulted in no platinum deposition in the polymer electrolyte membrane. These results suggest that platinum dissolved at a high potential in potential cycling and diffused into the membrane, and this was followed by deposition. Although Patterson described a band of platinum seen in the membrane after potential cycling for 3500 cycles based on an observation with electron probe microanalysis, the TEM technique used in this report can clearly detect even a small amount of platinum deposition in the membrane.15 Based on a careful analysis of the polymer electrolyte membrane using nano-EDX attached to the TEM, platinum was not detected in the region of the membrane where platinum particles did not appear, even in the region quite near the cathode interface. Therefore, the concentration of platinum ionic species dissolved in the electrolyte membrane is very low. Based on the TEM observation, it is probable that generation of the platinum nucleus is a rate-determining step in platinum-deposition. Platinum ionic species easily deposit on the platinum particles already in the polymer electrolyte and platinum particles grow using the platinum ionic species that have dissolved and diffused from the cathode catalyst layer. On the other hand, in the region near the anode catalyst layer, none of the samples showed deposited platinum particles in the polymer electrolyte by TEM observation. Platinum was seen only near the cathode by TEM observation. In studies that have examined platinum dissolution in aqueous electrolyte solution, the amount of platinum dissolved and diffused out of an electrode was determined by measuring the concentration of platinum in the electrolyte solution11–13 or the weight loss of the platinum electrodes.14 However, in the case of an MEA, it is difficult to measure the amount of dissolved platinum because a thin catalyst layer cannot be precisely peeled off or separated from the thin membrane after hot-pressing and dissolved platinum exists only near the interface between the catalyst layer and membrane.
 | ||
| Fig. 4 TEM images of the interface between the cathode catalyst layer and a polymer electrolyte membrane of an MEA after a potential cycling test for 500 cycles under a nitrogen atmosphere (samples N-1, N-2 and N-3). Potential cycling range: (a) 0.1–1.2 V, (b) 0.1–1.0 V, (c) 0.1–0.8 V vs. RHE. Membrane: Nafion® 117. | ||
 | ||
| Fig. 5 A typical TEM micrographic image of a deposited particle in a polymer electrolyte membrane near the cathode catalyst layer after a potential cycling experiment for 500 cycles under a nitrogen atmosphere (sample N-2). Potential cycling range: 0.1–1.0 V vs. RHE. | ||
The distribution of platinum deposited into the membrane after potential cycling ranging from 0.65 to 1.2 V (N-4) was similar to that in the case of 0.1 to 1.2 V (N-1, Fig. 4a), based on the results of TEM observations. Therefore, we think that the lower potential limit does not affect the platinum particle distribution in the polymer electrolyte membrane.
The dependence of platinum particle deposition in the membrane on the membrane thickness was investigated using a thinner membrane (Nafion® 112) with a thickness of 2 mil (50 μm). Potential cycling was carried out from 0.1 V to 1.2 V (N-5). When Nafion® 112 was used for the potential cycling experiment, platinum particle deposition in the electrolyte membrane was seen closer to the catalyst layer, as shown in Fig. 6 compared to Fig. 4a with Nafion® 117, which has a thickness of 7 mil (175 μm). This result suggests that some factor related to membrane thickness plays an important role in the platinum deposit distribution. In the MEA for PEMFC, it has been reported that some of the supplied gas such as hydrogen crosses the membrane.32,33 This “crossover” increases with decreasing membrane thickness since the concentration gradient within the membrane increases. As an example of the effect of crossover, Watanabe et al. proposed the “self-humidification” of PEMFC using platinum particles deposited in the membrane which catalyze the oxidation of crossover hydrogen with oxygen to generate water within the membrane.34 Therefore, it is considered that hydrogen from the anode (counter electrode) compartment through the membrane reduces platinum ionic species to platinum metal deposits and the distribution of platinum particles depends on the concentration of hydrogen in the membrane.
 | ||
| Fig. 6 A TEM image of the interface between the cathode catalyst layer and the polymer electrolyte membrane of an MEA after a potential cycling test for 500 cycles under a nitrogen atmosphere (sample N-5). Potential cycling range: 0.1–1.2 V vs. RHE. Membrane: Nafion® 112. | ||
Effects of hydrogen
To investigate the effects of hydrogen, potential cycling was carried out in the absence of hydrogen using cell B connected to a reference electrode by an electrolyte junction (N-6). Since this experiment was carried out with a three-electrode system, cell B, the anode potential was not regulated in a nitrogen atmosphere during the potential cycling experiment and rose to a higher potential than with a hydrogen atmosphere during cathodic scan of the cathode. MEA with an anode prepared from carbon black without platinum loading was used to prevent the effect of anode catalyst dissolution during the cathodic scan of the cathode.To more certainly prevent hydrogen generation, the lower potential limit was increased to 0.15 V vs. RHE. Platinum deposition was not seen in the resulting TEM image near the cathode (TEM image is not shown here). In addition, platinum particles were not seen either near the anode or in the membrane far from the electrodes based on the TEM observation of the sample. These results show that the reduction of platinum ions in a polymer electrolyte membrane was remarkably promoted by hydrogen from the anode compartment.
A typical electron micrograph of the anode catalyst layer near the electrolyte membrane is shown in Fig. 7. This figure shows platinum particles in the anode layer. Since this counter electrode contained no platinum before the experiment, it is clear that these platinum particles originate in the cathode. Platinum particles on the anode are considered to be deposited by the reduction of platinum ions on the carbon surface of the anode. Consequently, it was found that platinum ions can move across the electrolyte membrane from the cathode to the anode in the absence of hydrogen.
 | ||
| Fig. 7 A TEM image of the interface between the polymer electrolyte membrane and the anode catalyst layer of an MEA after a potential cycling test for 500 cycles under a nitrogen atmosphere using cell B connected to a reference electrode by an electrolyte junction (sample N-6). Potential cycling range: 0.1–1.2 V vs. RHE. Membrane: Nafion® 117. | ||
It is assumed that Pt2+ (or Pt4+) is the ionic platinum species in MEA under ideal conditions35 because a polymer electrolyte membrane is a non-complexing acid. Although it is generally thought that the platinum ion that does not form a complex, has low solubility and is readily reduced to platinum metal, platinum was found to be transported across the membrane even with a thicker membrane (Nafion® 117). If the ionic platinum species are an anionic complex made of chloride ion from some impurity or fluoride ion formed by membrane degradation,36 they may be more stable than cationic species. This result suggests that the optimized surface of an anode electrocatalyst, PtRu, may deteriorate due to the deposition of platinum from the cathode over most of the surface of PtRu if in the absence of hydrogen.
Effect of oxygen
To investigate the effect of oxygen on platinum deposition, potential cycling was carried out under air using cell A, without a reference electrode. The anode compartment was supplied with hydrogen. The potential was cycled between 0.65–1.2 V vs. RHE using the anode as a reference electrode (A-1). In this experiment, the lower potential limit was raised to 0.65 V, which is in the same range as in N-4.A TEM image of the cathode in sample A-1 is shown in Fig. 8. Platinum deposition was not observed near the cathode interface but was found about 20–25 μm further away. Fig. 9 compares the cyclic voltammograms under different atmospheres. Oxygen reduction current passes through the membrane in air. Hydrogen is oxidized at anode by this oxygen reduction current, resulting in a decrease in hydrogen permeation to the cathode because of hydrogen consumption at the anode catalyst layer. Therefore, the location at which the platinum ion is reduced can move toward the anode with a decrease in the hydrogen concentration in the membrane. In addition, we cannot entirely exclude the possibility that oxygen reduction current that passes through the membrane in air could affect the distribution of platinum if the ionic platinum species is an anionic complex that includes an impurity such as chloride or fluoride and moves with this current. However, the cathodic current in air is not much greater than that under a nitrogen atmosphere, as shown in Fig. 9. We are currently performing an experiment of long-term potential-holding at a high potential, 1.0 V, where oxygen reduction current does not occur, and so far we have found that platinum can deposit far from the cathode. Consequently, we think that the movement of platinum in this case was brought about by the diffusion of platinum ionic species and hydrogen consumption at the anode cannot account for the difference in platinum distribution in the membrane.
 | ||
| Fig. 8 TEM images of a cross-section of MEA after a potential cycling test for 500 cycles under an air atmosphere: (a) the region in a polymer electrolyte membrane about 20–25 μm from the cathode interface; (b) interface between the polymer electrolyte membrane and the cathode catalyst layer of MEA (sample A-1). Potential cycling range: 0.1–1.2 V vs. RHE. Membrane: Nafion® 117. | ||
 | ||
| Fig. 9 Comparison of the cyclic voltammograms under two different atmospheres (samples N-4 and A-1): (a) nitrogen, (b) air. Potential cycling range: 0.65–1.2 V vs. RHE. | ||
There are several possible explanations for this phenomenon. In a PEMFC, a small amount of the supplied oxygen can also transport across the membrane, and therefore there is a concentration gradient of oxygen as well as hydrogen across the polymer electrolyte membrane. In the case of an air atmospheric cathode, the region near the cathode interface has a high oxygen concentration and a low hydrogen concentration. Under these conditions, a platinum nucleus is not easily generated by reduction with hydrogen because a very small platinum nucleus might be unstable with high potential (mixed potential composed of dissolved hydrogen and oxygen) under such oxygen-rich conditions. Therefore, nucleus generation occurs more easily closer to the anode.
Platinum dissolution and deposition in the membrane are shown schematically in Fig. 10. The platinum ion dissolves at high potential and diffuses into the polymer electrolyte. That which diffuses out of the catalyst layer without redeposition on platinum catalyst particles is chemically reduced by hydrogen. The rate of the nucleus generation in the membrane is particularly accelerated by hydrogen. On the other hand, a high concentration of oxygen suppresses nucleus generation in the membrane. Consequently, nucleus generation depends on the concentration of oxygen as well as hydrogen in the membrane. Without hydrogen, the platinum ion deposits on the anode catalyst.
 | ||
| Fig. 10 Schematic drawing of platinum deposition in a polymer electrolyte membrane. | ||
Conclusion
Platinum dissolution and deposition in the polymer electrolyte membrane of an MEA for a PEMFC was studied using a potential cycling experiment and high-resolution transmission electron microscopy. TEM imaging of the MEA cross-section revealed platinum deposition in the polymer electrolyte membrane after potential cycling with an upper potential limit of 1.0 and 1.2 V vs. RHE. The distribution of platinum deposition was affected by the upper potential limit and membrane thickness. Hydrogen within the polymer electrolyte membrane promoted the formation of metallic aggregates. Platinum was transported across the membrane and deposited on the anode layer in the absence of hydrogen in the anode compartment. The distribution of platinum deposition was also affected by the presence of oxygen in the cathode compartment.Acknowledgements
This work was financially supported by the New Energy and Industrial Technology Development Organization (NEDO) as a part of the research and development of polymer electrolyte fuel cell technology project directed by the Ministry of Economy, Trade and Industry (METI). The authors thank Dr K. Tanaka, Mrs. N. Fujiwara and S. Yamazaki for their contributions to this work. The authors are grateful to Ms J. Maekawa for help with the TEM studies.References
- F. de Bruijn, Green Chem., 2005, 7, 132–150 RSC.
- H. Inaka, S. Sumi, K. Nishizaki, T. Tabata, A. Kataoka and H. Shinkai, J. Power Sources, 2002, 106, 60–67 CrossRef CAS.
- P. Costamagna and S. Srinivasan, J. Power Sources, 2001, 102, 253–269 CrossRef CAS.
- F. Preli, Fuel Cells, 2002, 2, 5–9 CrossRef CAS.
- C. K. Dyer, J. Power Sources, 2002, 106, 31–34 CrossRef CAS.
- K. Yasuda, Electrochemistry, 2002, 70, 630–634 Search PubMed.
- M. Fowler, R. F. Mann, J. C. Amphlett, B. A. Peppley, P. R. Roberge, 3: Fuel Cell Technology and Applications, in Handbook of Fuel Cells—Fundamentals, Technology and Applications, ed. W. Vielstich, A. Lamm and H. A. Gasteiger, John Wiley & Sons, 2003, pp. 663–667 Search PubMed.
- T. R. Ralph and M. P. Hogarth, Platinum Met. Rev., 2002, 46, 117–135 CAS.
- M. S. Wilson, F. H. Garzon, K. E. Sickafus and S. Gottesfeld, J. Electrochem. Soc., 1993, 140, 2872–2877 CAS.
- J. Xie, D. L. Wood III, K. L. More, P. Atanassov and R. L. Borup, J. Electrochem. Soc., 2005, 152, A1011–A1020 CrossRef.
- D. A. J. Rand and R. Woods, J. Electroanal. Chem., 1972, 35, 209–218 CrossRef CAS.
- D. C. Johnson, D. T. Napp and S. Bruckenstein, Electrochim. Acta, 1970, 15, 1493–1509 CrossRef CAS.
- K. Kinoshita, J. T. Lundquist and P. Stonehart, J. Electroanal. Chem., 1973, 48, 157–166 CrossRef CAS.
- K.-i Ota, S. Nishigori and N. Kamiya, J. Electroanal. Chem., 1988, 257, 205–215 CrossRef CAS.
- T. Patterson, Pre-Print Archive-American Institute of Chemical Engineers, Spring National Meeting, New Orleans, LA, 2002, pp. 313–318 Search PubMed.
- (a) M. Kohmoto, K. Totsuka, S. Hitomi, H. Yasuda, M. Yamachi and G. S. Yuasa, Tech. Report, 2004, 1, 23–27 Search PubMed; (b) S. Hitomi, Y. Sawada, M. Kohmoto, Y. Yasunaga, H. Yasuda, M. Yamachi, 2004 Fuel Cell Seminar Abstract, Fuel Cell, San Antonio, TX, 2004-000137, 2004 Search PubMed.
- P. Yu, M. Pemberton and P. Plasse, J. Power Sources, 2005, 144, 11–20 CrossRef CAS.
- National Institute of Advanced Industrial Science and Technology (AIST), The FY 2003 Progress Report for Polymer Electrolyte Fuel Cells, NEDO, 2003, to be published on the NEDO website (in Japanese), http://www.nedo.go.jp/database/index.html Search PubMed.
- K. Scott, W. M. Taama and P. Argyropoulos, J. Appl. Electrochem., 1998, 28, 1389–1397 CrossRef CAS.
- T. V. Nguyen, J. Electrochem. Soc., 1996, 143, L103–L105 CAS.
- M. Mizuhata, K. Yasuda, K. Oguro and H. Takenaka, Denki Kagaku oyubi Kogyo Butsuri Kagaku, 1996, 64, 692–698 Search PubMed.
- A. Taniguchi, T. Akita, K. Yasuda and Y. Miyazaki, J. Power Sources, 2004, 130, 42–49 CrossRef CAS.
- O. J. Murphy, G. Duncan and D. J. Manko, J. Power Sources, 1994, 47, 353–368 CrossRef CAS.
- H. Kumpulainen, T. Peltonen, U. Koponen, M. B. Ergelin, M. Valkiainen and M. Wasberg, VTT Tiedotteita, 2002, 2137, 1–28 Search PubMed.
- A. Antolini, J. Mater. Sci., 2003, 38, 2995–3005 CrossRef CAS.
- S. Mukerjee and S. Srinivasan, 2: Electrocatalysis, in Handbook of Fuel Cells—Fundamentals Technology and Applications, ed. W. Vielstich, A. Lamm and H. A. Gasteiger, John Wiley & Sons, 2003, pp. 502–519 Search PubMed.
- A. C. C. Tseung and S. C. Dhara, Electrochim. Acta, 1975, 20, 681–683 CrossRef CAS.
- A. Honji, T. Mori, K. Tamura and Y. Hishinuma, J. Electrochem. Soc., 1988, 135, 355–359 CAS.
- M. Watanabe, K. Tsurumi, T. Mizukami, T. Nakamura and P. Stonehart, J. Electrochem. Soc., 1994, 141, 2659–2668 CAS.
- K. F. Blurton, H. R. Kunz and D. R. Rutt, Electrochim. Acta, 1978, 23, 183–190 CrossRef CAS.
- J. A. Bett, K. Kinoshita and P. Stonehart, J. Catal., 1976, 41, 124–133 CrossRef CAS.
- K. Broka and P. Ekdunge, J. Appl. Electrochem., 1997, 27, 117–123 CrossRef CAS.
- S. Cleghorn, J. Kolde and W. Liu, 3: Fuel Cell Technology and Applications, in Handbook of Fuel Cells—Fundamentals Technology and Applications, ed. W. Vielstich, A. Lamm and H. A. Gasteiger, John Wiley & Sons, 2003, pp. 566–575 Search PubMed.
- M. Watanabe, H. Uchida, Y. Seki, M. Emori and P. Stonehart, J. Electrochem. Soc., 1996, 143, 3847–3852 CAS.
- R. M. Darling and J. P. Meyers, J. Electrochem. Soc., 2003, 150, A1523–A1527 CrossRef CAS.
- R. Baldwin, M. Pham, A. Leonida, J. McElroy and T. Nalette, J. Power Sources, 1990, 29, 399–412 CrossRef CAS.
| This journal is © the Owner Societies 2006 |