Subtle differences in structural transitions between poly-L- and poly-D-amino acids of equal length in water
Yosef
Scolnik
a,
Irina
Portnaya
b,
Uri
Cogan
b,
Saar
Tal
a,
Rachel
Haimovitz
a,
Mati
Fridkin
a,
Avshalom C.
Elitzur
c,
David W.
Deamer
d and
Meir
Shinitzky
*a
aWeizmann Institute of Science, Rehovot, 76100, Israel. E-mail: meir.shinitzky@weizmann.ac.il
bThe Technion, Haifa, 32000, Israel
cBar-Ilan University, Ramat Gan, 52900, Israel
dUniversity of California, Santa Cruz, CA 95064, USA
First published on 5th December 2005
Abstract
Mirror-image asymmetric molecules, i.e., chiral isomers or enantiomers, are classically considered as chemically identical. Recent studies, however, have indicated that parity violation by the nuclear weak force induces a tiny energy difference between chiral isomers. Upon combination with a massive amplification process, expansion of this difference to a detectable macroscopic level may be achieved. Yet, experimental tests of this possibility, where one enantiomer is compared to the other in solution, are hampered by the possible presence of undetectable impurities. In this study we have overcome this problem by comparing structural and dynamic features of synthetic D- and L-polyglutamic acid and polylysine molecules each of 24 identical residues. In these water-soluble polypeptides helix formation is an intramolecular autocatalytic process amplified by each turn, which is actually unaffected by low level of putative impurities in the solvent. The helix and random coil configurations and their transition were determined in this study by circular dichroism (CD) and isothermal titration calorimetry (ITC) in water and deuterium oxide. Distinct differences in structure and transition energies between the enantiomeric polypeptides were detected by both CD and ITC when dissolved in water. Intriguingly, these differences were by and large abolished in deuterium oxide. Our findings suggest that deviation from physical invariance between the D- and L-polyamino acids is induced in part by different hydration in water which is eliminated in deuterium oxide. Based on the recent findings by Tikhonov and Volkov (V. I. Tikhonov and A. A. Volkov, Science 2002, 296, 2363) we suggest that ortho-H2O, which constitutes 75% of bulk H2O, has a preferential affinity to L-enantiomers. Differential hydration of enantiomers may have played a role in the selection of L-amino acids by early forms of life.
Introduction
Space symmetry of physical laws asserts that in any macroscopic chemical or physical reaction, where achiral molecules are converted to chiral products, the system as a whole remains absolutely racemic. However, when an autocatalytic arm is associated with such a process, a slight excess of one enantiomer can be expanded considerably.1–3 In the autocatalytic process of crystallization, such an enantiomeric enhancement can be induced by mechanical agitation,4 stirring,5 or even β-irradiation.6 Furthermore, it is now widely accepted that chiral isomers are inherently at a slightly different energy state due to the parity violation of the electro-weak nuclear force (parity violation energy difference, PVED). The tiny excess of one enantiomer in a racemic mixture due to PVED can, in principle, be amplified by an external autocatalytic process to a level of detectable macroscopic difference.7,8 Whether such a process could explain the initial selection which led to the homochirality of amino acids, saccharides and nucleic acids in the biological realm, remains uncertain.9,10The above possibility was previously addressed by us in systems of supersaturated solutions of D- and L-tyrosine. D-tyrosine reached a saturated solution of lower concentration than L-tyrosine.11,12 Furthermore, crystallization of supersaturated DL-tyrosine in water led to enantiomeric enhancement of L-tyrosine in the saturated aqueous layer.11,12 Unexpectedly, the difference in the rate and level of crystallization between D- and L-tyrosine was almost fully abolished in D2O solutions.12 We interpreted these findings by proposing a dual function of PVED. In the first, it provides a slight excess of L-tyrosine which is amplified in the crystallization autocatalytic process, while in the other it induces a preferential interaction of ortho-H2O, which constitutes 75% of the bulk, with the L-enantiomer.12 Namely L-tyrosine is slightly more hydrophilic than D-tyrosine in H2O, but much less so in D2O, where spin isomers are of a much lower distinction.13
In an earlier study,14 similar trends were observed. Micelles of N-palmitoyl or N-stearoyl D- or L-serine in water displayed intense CD bands of opposite directions which are abolished by disruption of the micelles with ethanol. These specific CD bands were attributed to chiral surfaces where the serine head groups are arranged in a unique set of spines which are integrated to a chiral micellar surface.14 However, the absolute level of the CD band in the D-serine micelles was about 50% stronger than that of the enantiomeric L-serine micelles. This difference indicated a tighter correspondence between the residues on the surface of the D-serine micelles due to a lesser interaction with the surrounding aqueous layer.
Experimental verification of the intriguing assertion that enantiomers, like natural amino acids, are not fully identical, is hampered by the possibility that when comparing bulk systems the presence of impurities, even in undetectable concentrations, can tip the apparent macroscopic balance to an erroneous level. In the following study we have overruled this notorious problem by comparing structures and their transitions of two separate systems of water soluble enantiomeric polypeptides: poly L- and poly D-glutamic acid and poly L- and poly D-lysine, each of 24 identical residues. In these polypeptides α-helical and random coil configurations and their transitions involve intramolecular autocatalytic processes where low level of impurities in the solvent can be considered as negligible.
Polyglutamic acid and polylysine are water soluble polypeptides which undergo structural changes related to the degree of ionization of their side chains.15–17 When in the ionized state, these polypeptides are at an equilibrium among fluctuating unstructured conformations, generally termed as “random coil”. In the neutral state they assume a well-characterized α-helical structure which has a distinctive circular dichroism (CD) spectrum.18 These structures and their transitions were studied in this report.
Experimental
Peptide synthesis and analysis
Synthesis of L-(Lys)24-amide, D-(Lys)24-amide, L-(Glu)24-amide and D-(Glu)24-amide were carried out on ADVANCED CHEMTECH APEX 393 multiple peptide synthesizer (Louisville, KY), employing the α-N-fluorenylmethoxycarbonyl (Fmoc) strategy, following the available commercial protocols of the company. Peptide chain assembly was conducted on Rink Amide resin (Novabiochem Lauflingen, Switzerland). Fmoc-L-Lys(Boc)-OH, Fmoc-D-Lys(Boc)-OH , Fmoc-L-Glu(Boc)-OH and D-Glu(Boc)-OH (Novabiochem, Lauflingen) served as building blocks. The overall purity of these compounds was >98%, yet over 90% of the impurities were decomposed inactive materials, as assessed by HPLC. Opposite enantiomeric active residues were undetectable by the vendor19 and were therefore considered as negligible.Coupling of amino acid units was achieved by using, at each step, 4 equivalents of Fmoc-amino acid with 4 equivalents of benzotriazol-1-yl-oxy-tris-pyrolidinophosphonium hexafluorophosphate (PyBOP).20 Following detachment from the polymeric support and concomitant deprotection, by acidolysis with trifluoroacetic acid (TFA). Crude oligopeptides were purified to homogeneity by reversed phase HPLC on a semi-preparative silica C-18 column (250 × 10 mm; VYDAC). Elution was accomplished by a linear gradient established between 0.1% TFA in water and 0.1% TFA in 70% acetonitrile in water (v/v). The products were collected by lyophilization and analysed for purity on an analytical C-18 colunm (VYDAC) and on pre-packed Chromolith Performance RP-18e column (4.6 × 100 mm; Merck, Darmstadt) using the above linear gradient. HPLC-efluents were monitored at 220 nm. Molecular weights were ascertained by mass spectrometry (VG Tofspec; Laser Desorption Mass Spectrometry; Fison instruments, Manchester, UK). For all synthesized polypeptide used in this study the recorded mass spectrometry values were within 0.2% of the calculated molecular weight. Based on our analyses, we estimated intramolecular purity be higher than 99% for all four polypeptides.
Synthetic poly-DL-glutamic acid, poly-DL-lysine and poly-DL-alanine were obtained from Sigma and were used without further purification.
Circular dichroism (CD)
CD measurements were performed in a 1 mm quartz cuvette, with Aviv CD spectrometer model 202, at 25° C. Each scanning was repeated at least five consecutive times and averaged. Background scanning was recorded in parallel with the solvent alone and subtracted. Concentrations of the polypeptides were determined by optical density measurements at 212 nm in a 1 mm quartz cuvette taking an extinction coefficient of 1700 M−1 cm−1.15,16 The final pH of the solutions was adjusted with 0.01 M H3PO4, 0.01 M Na3PO4 and 0.1 M NaOH.Isothermal titration calorimetry (ITC)
Titration calorimetry measurements, at 30 °C, were performed with a VP-ITC calorimeter (Microcal Northampton, MA). The reaction cell (1.4301 ml) was filled with degassed Poly L or D glutamic acid or lysine solution, and the injection-stirrer syringe (0.289 ml) was loaded with 25 mM HCl solution or 100 mM NaOH solution. The concentration of the poly peptides in different experiments was in the range of 1.2–1.8 mM, taking care at each specific experiment to use essentially identical concentrations of the L and D samples. The concentration of the polymer solution was measured at 212 nm, using ε = 17001,2 M−1 cm−1. In one set of experiments the solvent (cell and syringe) was water, and in a second set of experiments the solvent was 80% D2O −20% H2O (v/v). Aliquots of the injectant, in 5 μl increments, were injected into the cell, and the heat flow was measured. The titration experiment consisted of 45 injections. The duration of an injection was 10 s, with 180 s equilibration time allowed between two consecutive injections.Data analysis
Data analysis was carried out with ORIGIN 5.0 software (MicroCal).Results and discussion
Circular dichroism
The CD spectrum of the right handed α-helix of poly-L-glutamic acid or poly-L-lysine is expected to be a mirror image of the spectrum of the left handed helices of their D-polypeptides under the same conditions.18 Furthermore, the energetics associated with helix formation or breaking is assumed to be identical for any pair of polypeptides of identical size composed of the D or the L enantiomers.21 However, parity violation energy differences (PVED) between chiral enantiomers,7–9 which are extremely small (∼10−17 eV), can in principle be increased to a detectable level when associated with an amplifying mechanism.9–11 Helix formation in such polypeptides is a typical autocatalytic process, where each turn enhances propagation to the next turn and beyond as the helix builds up. The energetics associated with helix formation in enantiomeric polypeptides, such as poly-L- or poly-D-glutamic acid, may be thus slightly different, a possibility which has far reaching implications.Poly-L-glutamic acid, poly-D-glutamic acid, poly-L-lysine and poly-D-lysine, each blocked at the carboxylic terminus as amide, and each of precisely 24 monomers [poly (L-Glu)24, poly (D-Glu)24, poly (L-Lys)24, and poly (D-Lys)24, respectively], were synthesized by solid phase stepwise addition of monomers, then isolated by preparative HPLC and analyzed (see Experimental). Their purity was the highest we could achieve. The precise molecular weight recorded for the polypeptides and the absence of opposite enantiomeric residues in the starting materials,19 led us to assess a purity of >99%. The CD spectra of poly (L-Glu)24 and poly (D-Glu)24 at their α-helix (measured at pH 2.5) and random-coil (measured at pH 10.5) configurations are presented in Figs. 1 and 2. As shown, the CD spectra in Fig. 1 are mirror images typical of right handed and left handed α-helices, respectively. Similar CD mirror image spectra were also recorded with poly (L-lys)24 and poly (D-Lys)24 in their helix region (recorded in 0.1 M NaOH, not shown). The identity of the absolute CD bands further supported the assessment of high purity of the studied polypeptides since any presence of a contaminant in the polypeptide matrix is expected to induce a marked effect on the helix formation reflected in the CD band. The CD spectra of the helices in H2O were stable for approximately 30 min and then gradually declined, probably due to aggregation and precipitation.
 | ||
| Fig. 1 CD spectra of poly (D-Glu)24 and poly (L-Glu)24 in water at pH 2.5. The spectra were recorded in a 1 mm path-length, at a concentration of 3 mM per residue. The spectra were reproducible up to 30 min after preparation of the solutions. Thereafter, the intensity of the bands slowly declined, probably due to aggregation. Measurements in D2O were impaired due to rapid precipitation of the polymers. | ||
 | ||
| Fig. 2 CD spectra of poly (D-Glu)24 and poly (L-Glu)24 at pH 10.5 in H2O and D2O. The insert represents the spectral difference between H2O and D2O. The spectra were recorded using a 1 mm path length, at a concentration of 3 mM per residue. | ||
In the random coil region of both sets of polypeptides, the CD spectra of the enantiomeric couples were not identical mirror images, as presented for the polyglutamic acid set in Fig. 2, indicating a net difference in the equilibrium state of their random coil conformations. As shown in Fig. 2, D2O markedly affected the CD spectrum of poly (L-Glu)24 but had a significantly smaller effect on the spectrum of poly (D-Glu)24. It is reasonable to assume that in this region, small differences in energy of the fluctuating conformations, which determine the equilibrium, could account for the observed deviation from mirror image spectra in the random coil region of poly (D-Glu)24 and poly (L-Glu)24. In the α-helix region, on the other hand, the energies associated with the hydration and the intramolecular hydrogen bonding are presumably much larger and could mask small energy differences between these peptides which appear to be identical in their CD spectra.
Isothermal titration calorimetry
The above suggestion could be tested by comparing the relative transition energies between α-helical and random coil configurations. We therefore conducted a detailed energetic determination of the helix-to-coil transition in these sets of poly peptides under isothermal conditions. Isothermal titration calorimetry (ITC) profiles at increments of decreasing pH were determined at 30 °C, either in H2O or in a 4∶1 (v/v) mixture of D2O and H2O. A summary of the data derived from the ITC profiles of poly(L-Glu)24 and poly(D-Glu)24 is presented in Table 1 and typical profiles are displayed in Fig. 3. The ITC profiles in Fig. 3 can be divided into three distinct regions related to the degree of ionization of the glutamic acid side chains: pH > 6, where the polypeptides retain an equilibrium among random coiled structures, pH ∼ 6–3, the range of transition to α-helix, and pH < 3, where the polypeptides are at their α-helix conformation. This classification was verified by CD scanning in a series of buffers of pH 2.5–10 which indicated α-helix-to-coil transition at pH ∼ 6 for both poly peptides (not shown).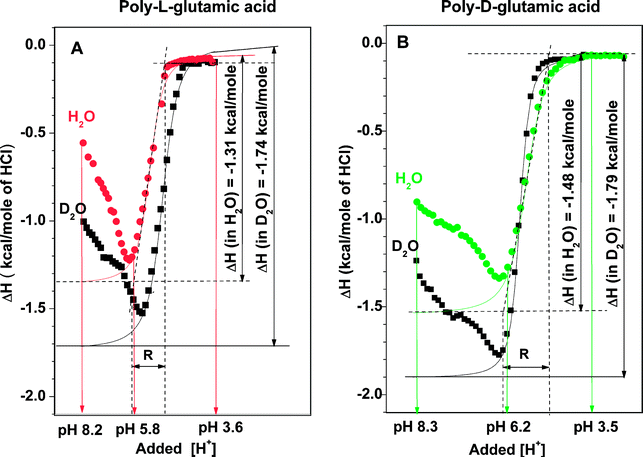
| In H2O | |||
|---|---|---|---|
| Poly(D-Glu)24 | Poly(L-Glu)24 | ||
| Experiment | ΔH/kcal (mol residue)−1 | ΔH/kcal (mol residue)−1 | ΔH difference/kcal (mol residue)−1 |
| a Derivation of cooperativity index is based on normalization, where the value of non-cooperativity equals 1.0 (see ref. 22). | |||
| 1 | −1.71 | −1.55 | −0.16 |
| 2 | −1.55 | −1.43 | −0.12 |
| 3 | −1.71 | −1.61 | −0.10 |
| 4 | −1.48 | −1.31 | −0.17 |
| Mean ± sd | −0.14 ± 0.03 | ||
| Transition pH | 6.2 | 5.8 | |
| Coil-helix | |||
| Transition midpoint | pH 5.8 | pH 5.0 | |
| Cooperativity indexa | 1.9 | 1.2 | |
| In mixture (D2O–H2O = 4 ∶ 1 v/v) | |||
| 1 | −1.88 | −1.95 | +0.07 |
| 2 | −1.79 | −1.74 | −0.05 |
| Mean (n = 2) | −1.84 | −1.85 | +0.01 |
In the α-helix region, the ITC profiles indicated marginal enthalpy change for both polypeptides. Similar ITC profiles of low enthalpy change were observed for acetic acid and propionic acid in water in the pH range of 2–8 (not shown), which suggests that the carboxylic side chains of poly(L-Glu)24 and poly(D-Glu)24 behave as isolated monomers at their α-helix region. On the other hand, in the random coil region (pH > 6) complex ITC profiles were recorded for both poly peptides, which indicated correspondence in enthalpy changes between the polypeptide backbone and the carboxylic head groups. The ITC profiles of the two enantiomeric polypeptides in this pH region were similar in shape but not identical (see Fig. 3), in agreement with the structural differences recorded by CD in this region (see Fig. 2).
The most pronounced differences between the ITC profiles of poly(L-Glu)24 and poly(D-Glu)24 in H2O were recorded in the transition region. The onset of the transition of poly(D-Glu)24 was 0.2–0.3 pH units above that of poly (L-Glu)24, namely, at a lower level of protonated side chains (see Table 1). This transition point is independent on concentration. In other words, the transition to α-helix of poly (D-Glu)24 started at a point of higher proportion of ionized side chains than in poly(L-Glu)24 (at pH 6.2 compared to 5.8, respectively), indicating a stronger tendency of poly-D-glutamic acid to adopt an α-helix structure. Both the degree of cooperativity and the associated change in enthalpy of the transition to α-helix of poly(D-Glu)24 were considerably higher than that of poly(L-Glu)24 (see Table 1). It is important to stress, that the abolition of the differences described above between the enantiomeric polypeptides in water by D2O, overrules the possibility of an undetectable flaw in their synthesis. Under such a hypothetical case the results in both solvents would be identical.
The most plausible interpretation for the above differences is that poly(D-Glu)24 has a higher α-helix stability than poly(L-Glu)24. According to this interpretation, each residue of D-glutamic acid in the turns which build the α-helix conformation, contributes approximately 0.1 kcal mol−1 more than that of L-glutamic acid in their enantiomeric helical turns. Taking 3.6 as the number of residues in each turn of α-helix, the excess energy of ∼0.4 kcal mol−1 turn in poly(D-Glu)24, is approximately equivalent to that of 10% of a hydrogen bond. In practice, ten α-helical turns of poly-D-glutamic acid would have an excess energy of an additional hydrogen bond compared to its poly-L-glutamic acid enantiomer. It is of great interest that in D2O–H2O 4 ∶ 1, the above differences were greatly diminished due to an almost selective effect on the ITC profile of poly (L-Glu)24 (see Fig. 3 and Table 1). This unexpectedly selective sensitivity of poly (L-Glu)24 to replacement of H2O by D2O, displayed in the CD spectra (Fig. 2) and the ITC profiles, provide the key issue in our suggested hypothesis which is presented below.
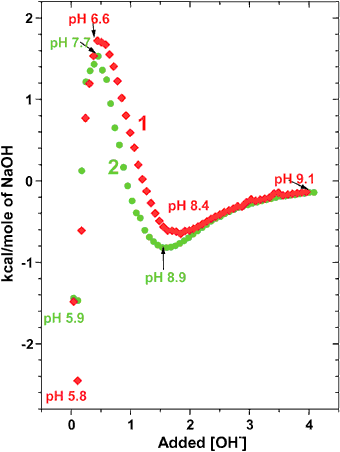
ITC profiles of poly(L-Lys)24 and poly(D-Lys)24 were measured and analyzed analogously to those presented above. The recorded ITC profiles of these polypeptides were qualitatively of a similar pattern to those shown in Fig. 3. Typical ITC profiles are presented in Fig. 4 and indicate that, unlike in the poly glutamic case, the transition to α-helix of poly(L-Lys)24 occurs at a higher degree of ionization than in poly(D-Lys)24 (pH 8.4 vs. pH 8.9, see Fig. 4).
Residual CD signal in poly-DL amino acids
The CD spectra presented in Fig. 1 and 2 suggest that synthetic poly-DL amino acids may display a small but non-zero CD signal. The random distribution of monomers in poly peptides made of synthetic DL amino acids implies that approximately 12% of the monomers reside in regions of four or more consecutive identical enantiomers (i.e., D,D,D,D… or L,L,L,L…). Assuming that the conformations in such regions comply with those observed in the enantiomeric polypeptides, then the net CD signal in poly amino acids may deviate from the expected complete annulation.This possibility was tested with commercial synthetic poly-DL-glutamic acid, poly-DL-lysine in the pH range of 4–10, as well as with poly-DL-alanine in water. A reproducible small CD signal of 0.5–0.8 mdeg at 221 nm was detected in poly-DL-glutamic acid at pH 5.5 and in poly-DL-lysine at pH 8.5 (see Figs. 3 and 4), as well as in poly-DL-alanine in water. However, these signals were at the lowest level of our instrumental resolution and were tentatively considered as inconclusive. It is interesting to note that along this line, Thiemann and Darge reported in 1974 on a ORD signal they observed in poly-DL-alanine.2
General discussion
The application of polymers for the detection of chiral deviations has two main advantages. The first relates to the notorious effects of impurities in tested solutions, which in polymers is reduced to defects in the polymer matrix which can be eliminated by building it from highly purified monomers. The other advantage corresponds to intramolecular autocatalytic amplification of a slight chiral deviation which propagates along the polymer backbone. An example of such amplification was found in polyisocyanates, where a single stereospecific deuterium substitution in the side chains was found to induce a strong CD signal corresponding to a unidirectional helix formation.23 The energy difference between the left- and right-handed helices in these polymers is extremely small and the specific deuterium substitution sufficed for tipping the perfect balance between the enantiomeric helices. Analogously, the energy difference in enantiomeric polypeptides, like those used in this study, is undoubtedly very small. As stated above, even such small energy difference could be amplified to a detectable level. The mechanism we suggest for the chiral amplification process in polypeptides is presented below.In the absence of an external discriminatory factor such as circularly polarized light, PVED remains the only physical effect which can lead to a selective and repetitive chiral enhancement.7–11 As PVED between isolated chiral isomers is extremely small, any expansion to the macroscopic realm must be associated with additional processes which lead to PVED amplification. In our poly-amino acid systems, amplification of PVED could, in principle, operate in two independent, yet cooperating, processes. The first is autocatalysis of helix formation or breaking, as clearly indicated in the ITC profiles of helix formation of our polypeptides (see Fig. 3 and 4 and Table 1). In the α-helix conformation of poly-amino acids, 3.6 residues constitute a complete turn. Since each turn contributes to the catalysis of helix formation, the 6–7 turns in poly (Glu)24 or poly (Lys)24 presumably correspond to a significant autocatalytic amplification of the inherent PVED of the monomers and could thus account in part for the observed differences in these sets of enantiomeric polypeptides.
The additional putative amplification arm is exclusive for aqueous solutions and corresponds to PVED induction of differences in the hydration layer between poly L-amino acids and their enantiomeric poly D-amino acids. In 2002 Tikhonov and Volkov24 reported an unexpected effect concerning proton exchange between the two spin isomers of H2O, namely ortho-H2O, where the proton spins are parallel, and para-H2O, where the proton spins are anti-parallel. They have shown that the exchange rates between these isomers are much slower than expected (half life of 30–50 min.). Further evidence for this unexpected slow exchange has been recently reported.25 One reasonable implication of this finding is that bulk water can be practically viewed as a mixture of ortho-H2O and para-H2O in a 3 ∶ 1 ratio (due to the three degenerate states of ortho-H2O). We have previously proposed12,26 that, since ortho-H2O bears a magnetic field, it has a slight preference to react with L-enantiomers due to their PVED induced magnetic component. As a result, in aqueous solutions of racemic mixtures, solvation preference of L-enantiomers may take place, which in the extreme cases may lead to chiral enhancement and further to a selective separation.12,26 In line with this hypothesis, a polypeptide of L-amino acids in water might be solvated slightly more than its mirror image poly-D-amino acids, so that the latter adopts an apparently more hydrophobic nature. In poly(L-Glu)24, poly(D-Glu)24, poly(L-Lys)24 and poly(D-Lys)24, where the asymmetrical conformations are determined almost exclusively by solvation energy and intramolecular hydrogen bonding, the structures and their transition energies in water could thus become significantly different.
If this hypothesis is correct, then the spin isomers of H2O and their putative selective effect on chiral isomers should be greatly diminished in D2O or D2O–H2O mixtures. Indeed, this is what we have found. In our ITC experiments we have used a mixture of D2O–H2O 4 ∶ 1 (v/v) as a system analogous to H2O but with “scrambled” spin isomers, namely, largely devoid of enantiomeric preference. In the steady state, this mixture is constituted of 4% H2O, 32% HDO and 64% D2O and from all physical and chemical aspects resembles pure H2O. As shown in Fig. 3, the pronounced differences in ITC profiles in H2O between poly(D-Glu)24 and poly(L-Glu)24 are by and large abolished in 80% D2O , as summarized in Table 1. Furthermore, the main effect of this mixture is on (L-Glu)24 and to a considerably smaller extent on poly(D-Glu)24. These observations, in addition to the differences presented in Fig. 2, comply with the above hypothesis that the preferred interaction of ortho-H2O with the L-enantiomer can account for a significant part of the structural and dynamic differences between enantiomeric polypeptides in water which were described in this study.
The slight difference in solvation between the enantiomeric polypeptides described above may have a considerable contribution to a difference in the entropy of transition, alongside the observed difference in the enthalpy of transition, as implied by the hierarchic model.27 The slightly less solvated poly-D-glutamic acid, may restrict the conformation balance in the random coil region, as compared with the poly-L-glutamic acid enantiomer. Therefore, the steering effects arising from hydrogen bonds and intramolecular interactions between residues in poly-D-glutamic acid may, by virtue of the relatively higher hydrophobicity of the D-isomer, be more pronounced. These will cause a higher restriction of the number of conformations, and create in the random coil a favorable backbone conformation of a higher tendency towards α-helix formation in the poly-D-glutamic acid.
Results indicating chiral discrimination between D- and L-alanine at their crystalline or aggregated states have already been provided by Wang et al.28–30
The results reported here imply that polypeptides comprised of D and L amino acids may have slightly different structures and transition energies than their mirror image enantiomeric polypeptides. This difference could be reflected in a difference in catalytic activity between such pairs of polypeptides, which may provide a clue for the selection of L-amino acids in the origin of life. In future work, this possibility could be tested with enantiomeric peptides which bear a catalytic site. This may clarify the thermodynamic advantage31 in the presumed chiral selection proposed in this study.
Acknowledgements
This research was supported in part by the Israel Science Foundation and by the Otto Meyerhof Center for Biotechnology at the Technion (Haifa), established by the Minerva Foundation (Munich, Germany).References
- H. J. Morovitz, J. Theor. Biol., 1969, 25, 491.
- W. Thiemann and W. Darge, Origins Life, 1974, 5, 263 CrossRef CAS.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767 CrossRef CAS.
- T. Buhse, D. Durand, D. Kondepudi, J. Laudadio and S. Silker, Phys. Rev. Lett., 2000, 84, 4405 CrossRef CAS.
- D. K. Kondepudi, R. J. Kaufman and N. Singhi, Science, 1990, 250, 975 CrossRef CAS.
- S. Mahurin, M. McGinnis, J. S. Bogard, L. D. Hulett, R. Pagni and R. N. Compton, Chirality, 2001, 13, 636 CrossRef CAS.
- R. A. Hegstrom, D. W. Rein and P. G. H. Sandars, Chem. Phys., 1980, 73, 2329 CrossRef CAS.
- S. F. Mason and G. E. Tranter, Mol. Phys., 1984, 53, 1091 CAS.
- A. Salam, J. Mol. Evol., 1991, 33, 105 CAS.
- W. A. Bonner, Orig. Life Evol. Biosphere, 1991, 21, 59 CAS.
- M. Shinitzky, F. Nudelman, Y. Bard, R. Haimoviz, E. Cohen and D. W. Deamer, Orig. Life Evol. Biosphere, 2002, 32, 285 CrossRef CAS.
- M. Shinitzky, A. C. Elitzur and D. W. Deamer, in Progress in Biological Chirality, ed. G. Playi, C. Zucchi and L. Caglioti, Elsevier, New York, 2004 Search PubMed.
- A. Farcas, Orthohydrogen, Parahydrogen and Heavy Hydrogen, Cambridge University Press, Cambridge, 1935 Search PubMed.
- M. Shinitzky and R. Haimovitz, J. Am. Chem. Soc., 1993, 115, 12545 CrossRef CAS.
- K. Imahori and J. Tanaka, J. Mol. Biol., 1959, 1, 359 CrossRef CAS.
- K. Rosenheck and P. Doty, Proc. Natl. Acad. Sci. USA, 1961, 46, 1775.
- T. Makovec, Biochem. Mol. Biol. Ed., 2000, 28, 244 Search PubMed.
- N. Greenfield and G. Fasman, Biochemistry, 1969, 8, 4108 CrossRef CAS.
- The optical purity examined by Nova Biochem was ≤0.5% while enantiomeric impurities were undetectable and were therefore assessed as <0.1%. The overall purity of the reacting compounds was assessed by HPLC to be higher than 99.5%.
- J. Martinez, J. Med. Chem., 1985, 28, 1874 CrossRef CAS.
- P. Doty and J. T. Yang, J. Am. Chem. Soc., 1956, 78, 498 CrossRef CAS.
- M. Shinitzky and M. Fridkin, Biochem. Biophys. Acta, 1976, 434, 137 CrossRef CAS.
- M. M. Green, J. W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem. Int. Ed., 1999, 38, 3138 CrossRef.
- V. I. Tikhonov and A. A. Volkov, Science, 2002, 296, 2363 CrossRef CAS.
- A. Miani and J. Tennyson, J. Chem. Phys., 2004, 120, 2732 CrossRef CAS.
- D. W. Deamer and M. Shinitzky, Astrobiology, 2006 Search PubMed , in press.
- R. L. Baldwin and B. H. Zimm, Proc. Natl. Acad. Sci. USA, 2000, 97, 12391 CrossRef CAS.
- W. Q. Wang, X. G. Sheng, H. F. Jin, J. Wu, B. Yin, J. Li, Z. X. Zhao, H. S. Yang, F. M. Lou and Z. Z. Zhuang, J. Biol. Phys., 1996, 22, 65 CrossRef CAS.
- W. Q. Wang, F. Yi, Y. Ni, Z. Zhao, Y. Jin and Y. Tang, J. Biol. Phys., 2000, 26, 51 CrossRef CAS.
- W. Q. Wang, Biophys. Chem., 2003, 103, 289 CrossRef CAS.
- A. C. Elitzur, J. Theor. Biol., 1995, 176, 349 CrossRef CAS.
| This journal is © the Owner Societies 2006 |