New tetrazine-based fluoroelectrochromic window; modulation of the fluorescence through applied potential†
Yuna
Kim
a,
Eunkyoung
Kim
*a,
Gilles
Clavier
b and
Pierre
Audebert
*b
aDepartment of Chemical Engineering, Yonsei University, 134 Shinchon-dong Seodaemun-gu, Seoul 120-749, South Korea. E-mail: eunkim@yonsei.ac.kr
bP. P. S. M., UMR 8531, Ecole Normale Supérieure de Cachan, 61 Av. du Pt Wilson, 94235 Cachan, France. E-mail: audebert@ppsm.ens-cachan.fr
First published on 26th July 2006
Abstract
A new electrofluorescent switch was prepared with an electroactive fluorescent tetrazine blend of polymer electrolyte; the cells contain four layers: the tetrazine polymer film, a photocured polymer electrolyte film, and two indium-tin oxide plates as the two contact electrodes.
Reversible electrochemistry of organic molecules accompanied by an optical change is an important issue in organic electronics such as electrochromic displays or electro-optical switches. While many electrochromic systems1 involving switchable chromophores included in or grafted on polymers have been described in the literature, there are very few reports of direct fluorescence modulation through the electrochemical potential applied to the fluorophore.2 Besides, there is no example of a fluorescent device where the fluorescence can be switched on and off by modifying the redox state of the fluorescent molecule itself, between its neutral state and one ion-radical state for example.
On the other hand, it was recently discovered that some tetrazine derivatives,3,4 besides being strongly fluorescent molecules, could also be electrochemically reduced to a stable anion-radical state in solution, in the classical conditions of analytical electrochemistry, i.e. using a microelectrode at concentrations in the 10−2–10−3 range. In addition, the tetrazines could also be reduced a second time to an unstable dianion state. Even though the dianion was transformed into some unstable species within a few seconds, the re-oxidation of this species regenerated the initial neutral tetrazine upon completion of the reverse scan.
These molecules therefore are very promising candidates to examine electrochemical fluorescence switching, in which the fluorescence of the neutral state could be reversibly switched on and off by converting the molecules successively to their reduced form (possibly non fluorescent) and back to the neutral (fluorescent) state. However, although the anion-radical was found to be stable on the timescale of cyclic voltammetry, no stable solution of the anion-radical could be obtained in order to check the fluorescence of this state. Attempted exhaustive electrolysis of tetrazine solutions gave rise to many by-products, probably arising from slow reactions with the solvent moisture.
However, we reasoned that in a thin layer cell the reduction could be fast enough to limit adventitious reactions, while converting all the tetrazines inside. We describe here the first example of a bilayer sandwiched polymeric device including tetrazines, where the fluorescence can be reversibly electrochemically switched on and off as a function of the applied potential.
The cell is a sandwiched device made of two layers packed between two transparent ITO electrodes (Fig. 1). It was prepared in two steps: first, a layer of a viscous polymer electrolyte solution was deposited cautiously on an ITO plate by spin coating and then cured so that it became a stiff film. Then a layer of the polymer electrolyte solution containing 5% weight of chloromethoxytetrazine was coated on the other ITO plate. Curing clearly improves the mechanical properties of the coating, but this was not possible with the tetrazine containing layer, because the UV irradiation resulted in the destruction of the tetrazine as seen by the loss of its distinctive red colour (see ESI for details).
 | ||
| Fig. 1 Diagram of the cell. | ||
The two plates were then contacted, taking care to avoid bubble formation at the interface, and firmly held together with two clips before connection to a standard potentiostat. The counter electrode and the reference electrode were connected together, so on the cyclic voltammograms (CV) it is the difference between the two electrode potentials which is registered (using a reference electrode was not possible with such a thin layer device).
Fig. 2 shows the CV for the tetrazine registered in the cell. While the formation of the anion-radical is slightly reversible, the formation of the dianion is not, but the starting tetrazine is restored at the end of the return sweep, as previously recognized in most cases of tetrazine electroreduction in solution. It is intriguing that the potentials for the tetrazine reduction are close to the ones registered in the case of analytical electrochemistry against an Ag/Ag+ reference electrode. This is due to the fact that in the case of the thin cell, the counter electrode reaction is very likely to be the oxidation of the titanocene produced by the decomposition of the photoinitiator Irgacure 784. Since titanocene is oxidized at around 0 V (vs. Ag/Ag+)5 the remaining potential difference (about 200 mV) probably arises from the ohmic drop in the cell.
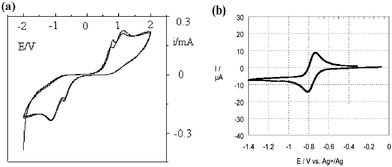 | ||
| Fig. 2 (a) Cyclic voltammogram of chloromethoxytetrazine in the thin cell and (b) standard CV of its first reduction in dichloromethane (from ref. 3). | ||
Fig. 3 displays a series of pictures showing the fluorescence of the device as a function of the applied electrochemical potential. It demonstrates that this device, which is fluorescent at the start (A), becomes less emissive when a negative potential is applied (B) and the emission is almost extinguished when the applied potential is −3 V (C). However, most of the fluorescence is restored on the return sweep (D).
 | ||
| Fig. 3 Successive images of the cell fluorescence recorded at different potentials, A: 0 V; B: −1.25 V; C: −3 V; and D: +1 V. | ||
This behaviour is also evidenced by the fluorescence spectra recorded at various potentials (Fig. 4a). It is clear that the fluorescence intensity is dependent upon the applied potential and almost reversibly extinct upon potential scanning toward negative values. Such electro-fluorescence switching was found to be reversible upon repetitive cycling between +2 V and −2 V (Fig. 4b). In this experiment, the potential was quickly scanned without looking for the full reduction of the tetrazines which explains why the variation of intensity is smaller than expected from the spectra. The cause of the feeble loss of overall intensity is still unclear and could come from some degradation of the fluorophore by photobleaching or the reaction of the anionic tetrazine, or from an incomplete reversed scanning. But the fluorescent contrast between the two states remains the same even after 120 cycles.
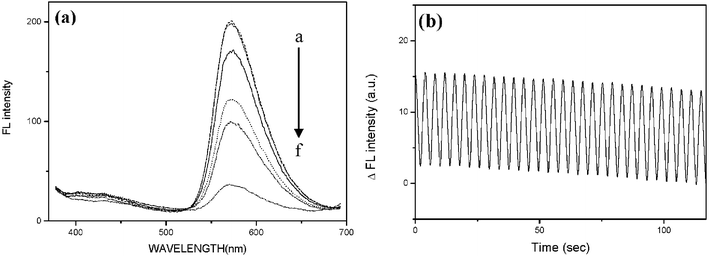 | ||
| Fig. 4 (a) Fluorescence spectra (λexc = 365 nm) of chloromethoxytetrazine recorded in the cell at different applied potentials; a: 1.25 V, b: 2 V, c: start, d: −1.25 V, e: −2 V, f: −3 V. (b) Fluorescence modulation of the window by potential switching from +2 V to −2 V, 2 s for each step. | ||
The fluorescent switching in the study originates from the reversible redox properties of tetrazines sandwiched between a photocured polymer electrolyte layer and an ITO electrode. It differs from conventional organic electroluminescence operating in LEC or LEDs.6–8 In LED, the semiconducting organic layer is oxidized on one side (holes are injected) and reduced on the other side (electrons are injected), but doping does not take place. LEC rely on transport of the oxidized or reduced light-emitting molecules (ions) themselves through the electrolyte between the electrodes. Unlike electrochromism, to our knowledge this is the first example where direct fluorescence switching can be monitored by electrochemical potential in an all solid state cell in which the fluorescent tetrazine molecules are in contact with the solid polymer electrolyte.
We have demonstrated the feasibility of the electrochemical switching of the emission of an intrinsically redox active fluorescent molecule into a multitime operated device. Such devices might find application in large size display panels operated using UV light. Work is in progess to optimise the cell response time and to improve the durability of the device.
Notes and references
- J. R. Platt, J. Chem. Phys., 1961, 34, 862 CrossRef CAS; M. Gratzel, Nature, 2001, 409, 575 CrossRef CAS; D. R. Rosseinsky and R. J. Mortimer, Adv. Mater., 2001, 13, 783 CrossRef CAS; A. A. Argun, P. H. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. MacDiarmid and J. R. Reynolds, Chem. Mater., 2004, 16, 4401 CrossRef CAS; R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2 CrossRef CAS; S. Jung, H. Kim, M. Han, Y. Kang and E. Kim, Mater. Sci. Eng., C, 2004, 24, 57 CrossRef; G. Sonmez, H. B. Sonmez, C. K. F. Shen, R. W. Jost, Y. Rubin and F. Wudl, Macromolecules, 2005, 38, 669 CrossRef CAS.
- Q. B. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086 CrossRef CAS; M. A. De Paoli and W. A. Gazotti, J. Braz. Chem. Soc., 2002, 13, 410; Q. B. Pei, Y. Yang, G. Yu, C. Zhang and A. J. Heeger, J. Am. Chem. Soc., 1996, 118, 3922 CrossRef CAS; F. G. Gao and A. J. Bard, J. Am. Chem. Soc., 2000, 122, 7426 CrossRef CAS; R. A. Illos, E. Harlev and S. Bittner, Tetrahedron Lett., 2005, 46, 8427 CrossRef CAS.
- P. Audebert, F. Miomandre, G. Clavier, M. C. Vernieres, S. Badre and R. Meallet-Renault, Chem.–Eur. J., 2005, 11, 5667 CrossRef CAS; P. Audebert, S. Sadki, F. Miomandre and G. Clavier, Electrochem. Commun., 2004, 6, 144 CrossRef CAS; P. Audebert, S. Sadki, F. Miomandre, G. Clavier, M. C. Vernieres, M. Saoud and P. Hapiot, New J. Chem., 2004, 28, 387 RSC.
- F. Guckel, A. H. Maki, F. A. Neugebauer, D. Schweitzer and H. Vogler, Chem. Phys., 1992, 164, 217 CrossRef.
- S. Back, G. Rheinwald and H. Lang, J. Organomet. Chem., 2000, 601, 93 CrossRef CAS.
- G. H. Brilmyer and A. J. Bard, J. Electrochem. Soc., 1980, 127, 104 CAS; I. Rubinstein and A. J. Bard, J. Am. Chem. Soc., 1980, 102, 6641 CrossRef; F. R. F. Fan, A. Mau and A. J. Bard, Chem. Phys. Lett., 1985, 116, 400 CrossRef CAS; J. E. Bartelt, S. M. Drew and R. M. Wightman, J. Electrochem. Soc., 1992, 139, 70 CAS.
- M. M. Richter, F. R. F. Fan, F. Klavetter, A. J. Heeger and A. J. Bard, Chem. Phys. Lett., 1994, 226, 115 CrossRef CAS; I. Ruff and L. Botar, J. Chem. Phys., 1985, 83, 1292 CrossRef CAS; L. Geng, R. A. Reed, M. H. Kim, T. T. Wooster, B. N. Oliver, J. Egekeze, R. T. Kennedy, J. W. Jorgenson, J. F. Parcher and R. W. Murray, J. Am. Chem. Soc., 1989, 111, 1614 CrossRef CAS; M. Watanabe, T. T. Wooster and R. W. Murray, J. Phys. Chem., 1991, 95, 4573 CrossRef CAS; M. Watanabe, H. Nagasaka, K. Sanui, N. Ogata and R. W. Murray, Electrochim. Acta, 1992, 37, 1521 CrossRef CAS; C. S. Velazquez, J. E. Hutchison and R. W. Murray, J. Am. Chem. Soc., 1993, 115, 7896 CrossRef CAS; M. Watanabe, H. Nagasaka and N. Ogata, J. Phys. Chem., 1995, 99, 12294 CrossRef CAS.
- A. R. Brown, D. D. C. Bradley, J. H. Burroughes, R. H. Friend, N. C. Greenham, P. L. Burn, A. B. Holmes and A. Kraft, Appl. Phys. Lett., 1992, 61, 2793 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: 10.1039/b608312a |
| This journal is © The Royal Society of Chemistry 2006 |