Coordination of oxovanadium(V) in an expanded porphyrin macrocycle†
Jonathan L.
Sessler
*,
Elisa
Tomat
and
Vincent M.
Lynch
Department of Chemistry and Biochemistry, 1 University Station, The University of Texas at Austin, Austin, TX, USA. E-mail: sessler@mail.utexas.edu; Fax: +1 512 471 7550; Tel: +1 512 471 5009
First published on 2nd August 2006
Abstract
The formation of a dioxovanadium(V) complex of an expanded porphyrin-type Schiff base macrocycle is reported; the tetrapyrrolic ligand undergoes a tautomeric shift which permits a bimodal recognition of the nonspherical cationic guest.
The manifold role of vanadium in biology and physiology1,2 continues to fuel the interest in its coordination chemistry. In fact, numerous high-valent vanadium compounds have been studied for their promising insulin-mimetic effects, anticancer activity and antiviral properties.3–6 Among these are vanadium(IV) porphyrins, which are currently being studied for their anti-HIV properties.7 Vanadium porphyrins have also attracted attention as so-called petroporphyrins (i.e., metalloporphyrins found in fossil fuels),8 and because they may be used potentially to access and study a range of different metal-centered oxidation states. These rather disparate interests are providing an incentive to make and study new vanadium complexes of porphyrins and porphyrin analogues.
Core-expanded porphyrin analogues9 are an especially promising class of receptors for coordination chemistry studies. These synthetic oligopyrrolic macrocycles provide a wealth of different ligand geometries, cavity sizes, and functionalities that allow for the stabilization of metal complexes not normally accessible by porphyrins. To date, a range of different expanded porphyrin metal complexes have been described. However, to the best of our knowledge, the coordination of vanadium in this type of macrocycle has not hitherto been reported.‡ Indeed, the coordination chemistry of early transition metals in expanded porphyrins is all but unexplored. In this communication we describe the preparation and characterization of an oxovanadium(V) complex of the Schiff base tetrapyrrolic macrocycle 1 and detail the role of an enamine–imine tautomerism of the ligand in the course of the key metalation reaction. We also provide solid state evidence for an unusual type of bimodal complexation, wherein the oxo cation is stabilized in part via hydrogen bonds to the expanded porphyrin receptor.
In previous work, macrocycle 1, a 22 π-electron non-aromatic expanded porphyrin, was found to form complexes with the high-valent actinide oxocations UO22+, NpO2+ and PuO2+.10 We thus sought to investigate the coordination of lighter, first row transition metal congeners of these heavy metal oxocations.
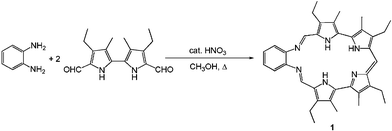
Since the first synthesis was reported in 1996,11 several analogues of macrocycle 1 have been prepared.10,12 The substitution pattern on the periphery of the macrocycle has been varied (on the pyrrolic rings, as well as on the phenyl ring and on the meso bridge), but all the reported synthetic strategies require the preparation of a diformylated tetrapyrrolic subunit prior to the cyclization with an ortho-phenylenediamine. We have now found a new efficient synthesis of this expanded porphyrin-type Schiff base macrocycle. It proceeds through a one-pot condensation/decarboxylation reaction and relies on the use of diformylbipyrrole as the only pyrrolic precursor (Scheme 1).
 | ||
| Scheme 1 | ||
The new synthesis of 1 relies on the reaction of diformylbipyrrole with 1,2-phenylenediamine. Similar condensations, carried out in the presence of catalytic HNO3, have been used in our laboratories for the preparation of a tetrapyrrolic octa-aza [2 + 2] Schiff base macrocycle.13 However, we have now found that the use of a 2 ∶ 1 diformylbipyrrole ∶ 1,2-phenylenediamine stoichiometry, along with slightly modified reaction conditions, directs the transformation towards the formation of the desired macrocycle 1 in 59% yield (see the Supporting Information for experimental details).§
A vanadium complex of expanded porphyrin 1 was obtained by treating the free base macrocycle with VO(acac)2 in THF at room temperature under aerobic conditions (yield: 61%). As shown in Fig. 1, the reaction was accompanied by a dramatic color change, meaning the conversion of the purple starting material 1 into the green vanadium complex 2 could be conveniently monitored by observing the changes in the UV-visible spectrum of the reaction mixture. After two days, the reaction was judged complete and the product was purified by preparative thin layer chromatography using alumina plates.¶
 | ||
| Fig. 1 UV-visible absorption spectra of macrocycle 1 (dashed line) and of the dioxovanadium(V) complex 2 (solid line) in CH2Cl2 at room temperature. | ||
Alternatively, complex 2 could be prepared using the V(V) reagent VO(O–iPr)3 under an argon atmosphere, but otherwise using the same purification procedure. However, when the initial metalation procedure, involving the use of VO(acac)2, was attempted under an inert atmosphere, no spectral evidence supporting the formation of a vanadium complex was observed. While trace quantities of product 2 were seen after a reaction time of four or five days, their formation was attributed to the presence of adventitious oxygen in the reaction vessel. In fact, while macrocycle 1 proved capable of stabilizing a dioxovanadium(V) complex, evidence for the formation of a vanadium(IV) complex could not be obtained.
The insertion of the dioxovanadium(V) cation was first inferred through electrospray ionization mass spectrometry. As often observed for metalation reactions using VO(acac)2 under aerobic conditions,14 the resulting complex features a vanadium center in its highest oxidation state. However, with six nitrogen donors available for coordination, no inferences could be drawn regarding the geometry of the VO2+ cation in complex 2.
An unambiguous description of the vanadium coordination geometry within the macrocycle cavity was obtained through X-ray diffraction analysis. Suitable single crystals grew as very dark lathes from a concentrated solution of 2 in CH2Cl2 and CH3OH at −20 °C.|| A top view of the refined crystal structure is shown in Fig. 2. The monovalent VO2+ cation, in the expected cisoidal conformation, coordinates to three pyrrolic nitrogen atoms acting as a tridentate monoanionic ligand. The vanadium center lies in a highly distorted trigonal bipyramidal environment with a geometric parameter τ of 0.57 (where τ values of 0 and 1 indicate an ideal square pyramid and an ideal trigonal bipyramid, respectively).15 The vanadium–oxygen distances (V1–O1, 1.618(3) Å and V1–O2, 1.635(3) Å), as well as the angle between the two oxo ligands (O1–V1–O2, 108.96(15)°), closely resemble those observed in other complexes of the VO2+ cation with a tridentate ligand.6,14,16 Similarly, the vanadium–nitrogen bond distances, averaging at 2.098 Å, compare well with those reported in the literature.6,16
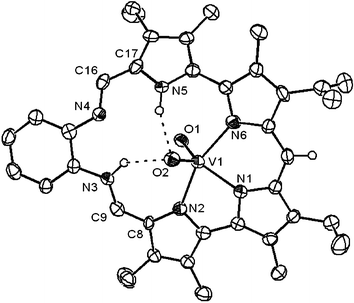 | ||
| Fig. 2 View of the dioxovanadium complex 2 showing a partial atom labeling scheme. Displacement ellipsoids are scaled to the 50% probability level. Most hydrogen atoms have been removed for clarity. A primary carbon on one of the ethyl groups is disordered about two orientations as shown. | ||
The structure of complex 2 can also be compared to those of vanadyl porphyrins, in which the N–V bond lengths typically range between 2.012 and 2.236 Å.17 However, it is important to appreciate that ligand 1 differs dramatically from porphyrin in that it supports the formation of a VO2+ complex, rather than a VO2+ complex, as always found in high oxidation state vanadium porphyrins.
Complex 2 also differs from porphyrins in that the ligand does not fully complex the bound cation. In fact, three of the six nitrogen atoms of the macrocycle do not participate in the metal coordination. The pyrrolic nitrogen N5 is engaged in a hydrogen bonding interaction with one of the oxo ligands (N5H⋯O2). Interestingly, the two remaining nitrogen atoms, bound to the phenyl ring, were found to be chemically different. A hydrogen atom bound to nitrogen N3 was located in a difference Fourier map. This hydrogen is involved in a second hydrogen bonding interaction with oxo ligand O2 (N3H⋯O2). Thus, while nitrogen N4 is iminic (N4–C16, 1.293(5) Å and C16–C17, 1.434(6) Å), nitrogen N3 was found to be part of an unusual enamine on the macrocycle skeleton (N3–C9, 1.330(5) Å and C9–C8, 1.385(6) Å).
In fact, the formation of the enamine tautomer 1′ (Scheme 2) alters the conjugation pathway of the macrocycle, thus allowing it to act as a monoanionic tridentate ligand for the vanadium center in complex 2. An ancillary effect of this rearrangement and metal coordination process is to engage one of the two oxo cation oxygen atoms in two hydrogen bonding interactions. As such, complex 2 represents a new contribution to the area of nonspherical guest recognition, namely one that features a bimodal binding between a metal oxocation and a single molecular receptor in which complexation of the nonspherical guest VO2+ is achieved through both covalent bonds and intramolecular hydrogen-bonding interactions.18,19
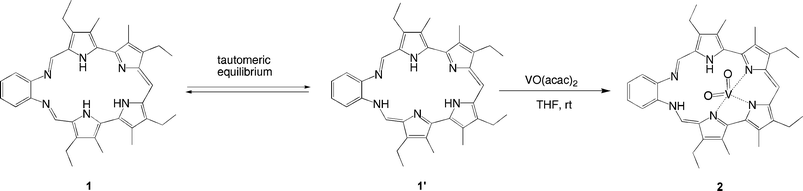 | ||
| Scheme 2 | ||
The room-temperature 1H NMR spectrum of complex 2 provides support for the notion that the solid state structure shown in Fig. 2 is retained in solution. As a result of the loss of symmetry in complex 2 compared to the free base macrocycle 1, the protons on the four pyrrolic methyl groups resonate as four different singlets in the aliphatic region. Additionally, nine different protons were found to resonate down-field between 7.5 and 11.5 ppm. Resonances in this region include: (a) a group of peaks in the 7.6–8.3 ppm range for the four expected aromatic protons and the proton on the meso bridge, (b) two doublets for the enaminic protons (a sharp CH peak at δH 8.81 ppm and a broad NH peak at 9.80 ppm), and (c) two singlets accounting for one iminic proton (δH 9.61 ppm) and one pyrrolic NH (δH 11.43 ppm).
In summary, we have developed a new synthesis for the Schiff base tetrapyrrolic macrocycle 1 and we have used this ligand to prepare an unprecedented dioxovanadium(V) expanded porphyrin complex. The elucidation of the VO2+ coordination mode leads to the conclusion that the macrocycle undergoes an imine–enamine tautomerism so as to maximize the number of interactions (both covalent and non-covalent) towards the singly charged nonspherical cationic guest. While this specific rearrangement is new in our experience, previous studies of Schiff base oligopyrrole macrocycles have provided several examples where differing cation coordination gerometries and structures are stabilized as the result of what appear to be ostensibly small changes in the charge and size of the metal cation, the protonation state of the metal-free macrocycle, and the nature of the counterions. Thus, the present work, which adds the presence of possible tautomeric equilibria to this list of factors, serves to highlight further the remarkable versatility of this class of ligands.20
This work was supported by the DOE (grant DE-FG02-01ER-15186 to J.L.S.).
Notes and references
- Metal Ions in Biological Systems, ed. H. Sigel and A. Sigel, Marcel Dekker, New York, 1995, vol. 31 Search PubMed.
- D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849–902 CrossRef CAS.
- (a) K. H. Thompson and C. Orvig, Coord. Chem. Rev., 2001, 219, 1033–1053 CrossRef; (b) K. H. Thompson, J. H. McNeil and C. Orvig, Chem. Rev., 1999, 99, 2561 CrossRef CAS.
- D. Rehder, G. Santoni, G. M. Licini, C. Schulzke and B. Meier, Coord. Chem. Rev., 2003, 237, 53–63 CrossRef CAS.
- (a) R. K. Narla, Y. H. Dong, O. J. D'Cruz, C. Navara and F. M. Uckun, Clin. Cancer Res., 2000, 6, 1546 CAS; (b) O. J. D'Cruz, Y. H. Dong and F. M. Uckun, Biochem. Biophys. Res. Commun., 2003, 302, 253–264 CrossRef CAS.
- P. Noblío, M. Vieites, B. S. Prarajón-Costa, E. J. Baran, H. Cerecetto, P. Draper, M. Gonzáles, O. Piro, E. E. Castellano, A. Azqueta López de Ceráin, A. Monge-Vega and D. Gambino, J. Inorg. Biochem., 2005, 99, 443–451 CrossRef CAS.
- S.-Y. Wong, R. W.-Y. Sun, N. P.-Y. Chung, C.-L. Lin and C.-M. Che, Chem. Commun., 2005, 3544–3546 RSC.
- H. Xu, G. Que, D. Yu and J. R. Lu, Energy Fuels, 2005, 19, 517–524 CrossRef CAS.
- (a) J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134–5175 CrossRef CAS; (b) W. B. Callaway, J. M. Veauthier and J. L. Sessler, J. Porphyrins Phthalocyanines, 2004, 8, 1–25 CrossRef CAS.
- J. L. Sessler, A. E. V. Gorden, D. Seidel, S. Hannah, V. M. Lynch, P. L. Gordon, R. J. Donohoe, C. D. Tait and D. W. Keogh, Inorg. Chim. Acta, 2002, 341, 54–70 CrossRef CAS.
- J. L. Sessler, V. Král, M. C. Hoehner, K. O. A. Chin and R. M. Dávila, Pure Appl. Chem., 1996, 68, 1291–1295 CrossRef CAS.
- (a) A. Drews, T. Schönemeier, S. Seggewies and E. Breitmaier, Synthesis, 1998, 749–752 CrossRef CAS; (b) S. Meyer, M. C. Hoehner, V. M. Lynch and J. L. Sessler, J. Porphyrins Phthalocyanines, 1999, 3, 148–158 CrossRef CAS.
- J. L. Sessler, T. D. Mody and V. M. Lynch, J. Am. Chem. Soc., 1993, 115, 3346 CrossRef CAS.
- M. R. Maurya, Coord. Chem. Rev., 2003, 237, 163–181 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- (a) P. Plitt, H. Pritzkow and R. Krämer, Dalton Trans., 2004, 2314–2320 RSC; (b) M.-J. Xie, Y.-S. Ping, L.-D. Zheng, J.-Z. Hui and C. Peng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1382–m1383 CrossRef; (c) A. G. J. Ligtenbarg, A. Speck, R. Hage and B. L. Feringa, J. Chem. Soc., Dalton Trans., 1999, 659–661 RSC.
- (a) R. Guilard and C. Lecomte, Coord. Chem. Rev., 1985, 65, 87–113 CrossRef CAS; (b) F. S. Molinaro and J. A. Ibers, Inorg. Chem., 1976, 15, 2278–2283 CrossRef CAS; (c) D. Afzal, R. Baughman, A. James and M. Westmeyer, Supramol. Chem., 1996, 6, 395–399 CrossRef CAS; (d) R. Harada, H. Okawa and T. Kojima, Inorg. Chim. Acta, 2005, 358, 489–496 CrossRef CAS.
- For early examples of multi-mode coordination involving, respectively, the dioxouranium(VI) and the dioxoosmium(VI) cations, see: (a) T. S. Franczyk, K. R. Czerwinski and K. N. Raymond, J. Am. Chem. Soc., 1992, 114, 8138–8146 CrossRef CAS; (b) A. S. Borovik, J. Du Bois and K. N. Raymond, Angew. Chem., Int. Ed. Engl., 1995, 34, 1358–1362.
- For examples of multi-mode stabilization of the uranyl cation in an oligopyrrolic macrocycle, see: (a) P. L. Arnold, A. J. Blake, C. Wilson and J. B. Love, Inorg. Chem., 2004, 43, 8206 CrossRef CAS; (b) P. L. Arnold, D. Patel, A. J. Blake, C. Wilson and J. B. Love, J. Am. Chem. Soc., 2006, 128, 9610–9611 CrossRef CAS.
- (a) J. M. Veauthier, W.-S. Cho, V. M. Lynch and J. L. Sessler, Inorg. Chem., 2004, 43, 1220–1228 CrossRef CAS; (b) J. L. Sessler, E. Tomat, T. D. Mody, V. M. Lynch, J. M. Veauthier, U. Mirsaidov and J. T. Markert, Inorg. Chem., 2005, 44, 2125–2127 CrossRef CAS; (c) J. M. Veauthier, E. Tomat, V. M. Lynch, J. L. Sessler, U. Mirsaidov and J. T. Markert, Inorg. Chem., 2005, 44, 6736–6743 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, X-ray structural data for 2. See DOI: 10.1039/b608143f |
| ‡ Interesting vanadium complexes of non core-expanded artificial porphyrin derivatives have been reported. See: (a) J. Jubb, L. Scoles, H. Jenckins and S. Gambarotta, Chem.–Eur. J., 1996, 2, 767–771; (b) T. Fukuda, E. A. Makarova, E. A. Luk'yanets and N. Kobayashi, Chem.–Eur. J., 2004, 10, 117–133. |
| § Characterization data for macrocycle 1: (Found: C, 75.88; H, 7.68; N, 14.05. Calc. for [C37H42N6]·(H2O): C, 75.48; H, 7.53; N, 14.27%); λmax(CH2Cl2)/nm 385 (log ε 4.87), 580 (4.39); δH (400 MHz, 296 K, CD2Cl2) 8.65 (2 H, s, iminic CHN), 7.57–7.55 (2 H, m, ArH), 7.41–7.39 (2 H, m, ArH), 7.15 (1 H, s, meso), 2.82–2.75 (8 H, m, CH2CH3), 2.30 (6 H, s, CH3), 2.24 (6 H, s, CH3), 1.29 (6 H, t, CH2CH3), 1.23 (6 H, t, CH2CH3), (pyrrolic NH not observed). δC (100 MHz, 296 K, CD2Cl2) 147.4, 145.7, 144.4, 143.0, 138.4, 134.1, 129.3, 126.2, 124.5, 119.4, 117.3, 115.7, 18.6, 17.8, 16.7, 16.4, 11.8, 11.5; m/z (CI) 571.35650 ([M + 1]+ C37H43N6 requires 571.35437). |
| ¶ Characterization data for complex 2: (Found: C, 61.77; H, 5.60; N, 11.11. Calc. for [C37H41N6O2V]·(CH2Cl2): C, 61.87; H, 5.88; N, 11.39%); λmax(CH2Cl2)/nm 368 (log ε 4.83), 450 (4.87), 665 (4.19), 727 (4.58); δH (400 MHz, 296 K, CD2Cl2) 11.43 (1 H, s, pyrrolic NH), 9.80 (1 H, br d, ArNH), 9.61 (1 H, s, iminic CHN), 8.82 (1 H, d, CHNH), 8.24 (1 H, d, ArH), 8.22 (1 H, d, ArH), 7.92 (1 H, s, meso), 7.69–7.61 (2 H, m, ArH), 3.12–2.92 (8 H, m, CH2CH3), 2.77 (3 H, s, CH3), 2.70 (3 H, s, CH3), 2.50 (3 H, s, CH3), 2.31 (3 H, s, CH3), 1.51–1.27 (12 H, m, CH2CH3). δC (100 MHz, 296 K, CD2Cl2) 153.0, 150.0, 149.5, 149.0, 146.2, 139.6, 138.7, 136.7, 132.3, 131.8, 130.3, 129.8, 129.4, 126.4, 125.4, 123.7, 121.3, 118.8, 117.2, 114.1, 95.2, 19.5, 19.0, 18.9, 18.3, 17.2, 17.0, 16.8, 16.5, 13.5, 12.7; m/z (ESI) 653.2803 ([M + 1]+ C37H42N6O2V requires 653.2809). |
|| Crystal data for 2·(CH2Cl2)3: C40H47Cl6N6O2V, M
= 907.48, triclinic, a
= 12.4914(2)
Å, b
= 13.0026(3)
Å, c
= 14.8379(5)
Å, α
= 89.426(1)°, β
= 88.093(1)°, γ
= 61.536(1)°, U
= 2117.44(9)
Å3, T
= 153(2) K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z
= 2, λ(Mo-Kα)
= 0.71073 Å, μ
= 0.656 mm−1, crystal size 0.24 × 0.23 × 0.18 mm, 16243 reflections collected, 9540 unique (Rint
= 0.1101). The final wR(F2) was 0.1355 (all data). CCDC 610169. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b608143f , Z
= 2, λ(Mo-Kα)
= 0.71073 Å, μ
= 0.656 mm−1, crystal size 0.24 × 0.23 × 0.18 mm, 16243 reflections collected, 9540 unique (Rint
= 0.1101). The final wR(F2) was 0.1355 (all data). CCDC 610169. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b608143f |
| This journal is © The Royal Society of Chemistry 2006 |