Introduction of single mutation changes arylmalonate decarboxylase to racemase†
Yosuke
Terao
,
Kenji
Miyamoto
and
Hiromichi
Ohta
*
Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama, 223-8522, Japan. E-mail: hohta@bio.keio.ac.jp; Fax: +81-45-566-1551; Tel: +81-45-566-1703
First published on 3rd August 2006
Abstract
The introduction of only one mutation based on the estimated reaction mechanism endowed arylmalonate decarboxylase with a racemase activity, which catalyses racemisation of α-arylpropionates.
Owing to the recent development of biotechnology and informatics as well as databases, changing the function of enzymes has become possible via site-directed mutagenesis and directed evolution.1–4 Most of the changes are in the range of the same type of reaction, such as changing the substrate- and enantio-selectivity, increasing the activity and thermal stability.5 On the other hand, a small number of mutations directed to promiscuous enzymes have also been reported recently.6–8 In these cases, a mutated lipase and some acylases have been demonstrated to catalyse the aldol reaction, Michael addition, and Markovnikov addition of N-heterocycles to vinyl esters. In nature also, some enzymes have catalytic promiscuity with regard to different types of substrates.9 Such divergent evolution is considered to be at least one of the ways in which nature has achieved a wide variety of enzymes that catalyse a number of biotransformations.10 Especially, the interplay of functions and structures of enzymes belonging to the enolase superfamily has been extensively studied by Gerlt's and Rayment's groups.11–14 Some enzymes are sterically superimposable with each other, nonetheless they catalyse distinct reactions. Because the intermediate of the reaction catalysed by our enzyme described in this paper is estimated to be the enolate of carboxylic acid similarly to the case of the enolate superfamily, we expected that a simple mutation might endow the enzyme with new catalytic activity. In such a trial, consideration of the reaction mechanism and well-designed mutagenesis will be essential. Here, we would like to demonstrate that introduction of a single mutation changed the decarboxylase of malonates to the racemase of α-substituted monobasic acids.
We have been investigating a novel and unique enzyme, arylmalonate decarboxylase (AMDase, E.C. 4.1.1.76, originating from Alcaligenesis bronchisepticus KU1201), which catalyses enantioselective decarboxylation of α-aryl-α-methylmalonates to give optically active α-arylpropionates of high enantiomeric excess in high yields (Scheme 1).15,16 The gene encoding AMDase has already been cloned, overexpressed in E. coli, and the enzyme has been purified.17–19 The essential amino acid in the active site of AMDase is revealed to be Cys188 which is considered to protonate the intermediate enolate form of α-arylpropionates from only one side of the enantiomeric face to give (R)-products.20,21 AMDase has some homology with some racemases and isomerases, such as glutamate racemase,22 aspartate racemase,23 and others (Fig. 1).24,25 These enzymes are known to be categorized into cofactor-independent racemase and isomerase families.14 The common points between AMDase and these enzymes are as follows: the intermediates are the enolate form of the products and the essential amino acid residue in the active site is Cys188, which delivers a proton to the intermediate enolate. On the other hand, the marked difference between these enzymes is that while racemases have another Cys located in the opposite side of the enantiomeric face of the intermediates, AMDase has no corresponding Cys around this region. Thus, racemases can racemise the substrates via a two-base mechanism as revealed by analysis of the crystal structure and the site-directed mutagenesis,2,26–28 while AMDase gives the optically active decarboxylated products.
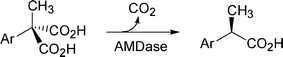 | ||
| Scheme 1 Asymmetric decarboxylation. | ||
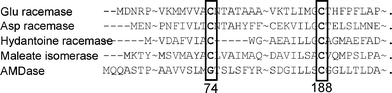 | ||
| Fig. 1 Amino acid homology between some racemases and AMDase. | ||
Based on the homology alignment we have prepared a double mutant AMDase (G74C/C188S) and confirmed that it gave the opposite enantiomer of arylpropionate compared to that obtained with the wild type enzyme.29
Now we would like to examine a further challenging idea. The above mentioned reactivity of G74C/C188S mutant led us to suppose that G74C single mutant might exhibit racemisation activity towards arylpropionates, in addition to its original decarboxylase activity.
Although decarboxylation and racemisation are entirely different reactions, the key intermediate is the same type of species, i.e., the enolate form of an α-substituted carboxylic acid. Encouraged by this fact, we introduced a G74C mutation into a wild type AMDase gene by site-directed mutagenesis. The E. coli JM109 was transformed with the plasmid containing G74C mutation and incubated in LB medium. The overexpressed G74C AMDase was purified in the same way as wild type enzyme.19
First, we examined the racemisation reaction of α-(2-naphthyl)propionic acid (2). The corresponding malonate is one of the best-accepted substrates of the decarboxylation reaction of wild type AMDase. Enantiopure 2 was prepared by our unique enzymatic decarboxylation reaction from the corresponding arylmalonate. Wild type AMDase gave (R)-2 with high enantiomeric excess, and (S)-2 was obtained with the aid of G74C/C188S mutant which was prepared recently.29
The racemization reaction was performed as follows. To a solution of (R)-2 or (S)-2 in Tris-HCl buffer (100 mM, pH 8.5) was added purified G74C mutant AMDase and the mixture was incubated at 37 °C for 14 h. After quenching the reaction with 2 M HCl, the product was extracted and its ee was measured by HPLC. As expected, G74C mutant AMDase gave the racemic product regardless of the configuration of the starting materials, while the control reaction without the enzyme resulted in no change in ee. Moreover, non-mutated wild type AMDase exhibited no racemase activity. These results indicate that the introduction of only one mutation, i.e., cysteine instead of glycine74, changed the decarboxylation enzyme to a racemase. The presence of two active cysteine residues probably enabled the two-base mechanism to work similarly to glutamate racemase.26–28
Next, we examined the substrate specificity of racemase activity using several compounds as shown in Table 1. In general, good substrates for decarboxylation reaction were also good substrates for racemisation. The most active group consisted of arylpropionates, such as α-phenyl- (1), α-(2-naphthyl)- (2), and α-(2-thienyl)propionic acid (3), which were followed by mandelic acid (5) and α-phenylbutyric acid (6). On the other hand, the rate of racemisation of phenylglycine (4) was very slow. The order of reactivity of these compounds is consistent with that of decarboxylation reactions of the corresponding malonates catalysed by the wild type AMDase. On the other hand, carboxylic acids which had larger α-substituents such as iso-propyl (7) or n-propyl (8) were inactive similarly to the case of decarboxylation. This is considered to be due to the limitation of the size of the active site pocket. The similarity of substrate specificity between decarboxylation and racemisation indicates that both reactions are catalysed by the same site of wild type and mutant enzymes. Carboxylic acid derivatives, such as alcohol (9), amide (10), nitrile (11), and ester (12), were totally inactive, indicating that the presence of a free carboxyl group is indispensable for G74C AMDase. The presence of an α-aryl group is also demonstrated to be essential for the substrates. It is worth noting that α-ethyl compound (6) was also racemised although the reactivity was relatively small (Table 1, entry 6). This is the only example that exhibited racemisation activity in spite of the inactivity of the corresponding malonate to decarboxylation reaction.
|
|
|||||
|---|---|---|---|---|---|
| Substrate | Ar | R | X | k cat/Km (s−1/mM) | Relative activity |
| 1 | Ph | Me | CO2H | 0.077 | 100 |
| 2 | 2-Np | Me | CO2H | 0.56 | 720 |
| 3 | 2-Th | Me | CO2H | 0.27 | 350 |
| 4 | Ph | NH2 | CO2H | 0.0038 | 5 |
| 5 | Ph | OH | CO2H | 0.011 | 14 |
| 6 | Ph | Et | CO2H | 0.018 | 23 |
| 7 | Ph | i-Pr | CO2H | — | 0 |
| 8 | Ph | n-Pr | CO2H | — | 0 |
| 9 | Ph | Me | CH2OH | — | 0 |
| 10 | Ph | Me | CONH2 | — | 0 |
| 11 | Ph | Me | CN | — | 0 |
| 12 | Ph | Me | CO2Me | — | 0 |

G74C mutant AMDase retained its original decarboxylase activity, and gave racemic arylpropionates when arylmalonates were employed as the substrates. There are two possible explanations for the formation of racemic products. One is racemisation of originally resulting optically active products and the other is the direct formation of racemic monobasic acids. To distinguish between these two mechanisms, the kinetic parameters of decarboxylation and racemisation were measured. It was revealed that the catalytic efficiency (kcat/Km) of racemisation of naphthylpropionate (2) is smaller (0.56 s−1 mM−1) than that of decarboxylation (0.96 s−1 mM−1). In addition, the decarboxylated product at the very initial stage of the reaction was confirmed to be racemic. These results indicate that G74C mutant decarboxylates the substrates non-selectively, delivering a proton from both sides of the intermediate enolate. Thus, introduction of one acidic amino acid residue brought about the protonation activity from both sides of the intermediate and gave the enzyme the racemisation activity. To the best of our knowledge, this is the first report of changing a decarboxylase to a racemase by the introduction of only one mutation based on the reaction mechanism.
This research was supported in part by the Ministry of Education, Culture, Sports, Science and Technology, Grant-in-Aid for the 21st Century Center of Excellence (COE) Program entitled “Understanding and Control of Life's Function via Systems Biology (Keio University)”. Financial support from Takeda Science Foundation is also gratefully acknowledged.
Notes and references
- A. Skandalis, L. P. Encell and L. A. Loeb, Chem. Biol., 1997, 4, 889 CrossRef CAS.
- M. E. Tanner, Acc. Chem. Res., 2002, 35, 237 CrossRef CAS.
- A. O. Magnusson, M. Takwa, A. Hamberg and K. Hult, Angew. Chem., Int. Ed., 2005, 44, 4582 CrossRef CAS.
- G. J. Williams, S. Domann, A. Nelson and A. Berry, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3143 CrossRef CAS.
- K. L. Morley and R. J. Kazlauskas, Trends Biotechnol., 2005, 23, 231 CrossRef CAS.
- M. Svedendahl, K. Hult and P. Berglund, J. Am. Chem. Soc., 2005, 127, 17988 CrossRef CAS.
- W.-B. Wu, J.-M. Xu, Q. Wu, D.-S. Lv and X.-F. Lin, Adv. Synth. Catal., 2006, 348, 487 CrossRef CAS.
- C. Branneby, P. Carlqvist, K. Hult, T. Brinck and P. Berglund, J. Mol. Catal. B: Enzym., 2004, 31, 123 CrossRef CAS.
- U. T. Bornscheuer and R. J. Kazlauskas, Angew. Chem., Int. Ed., 2004, 43, 6032 CrossRef CAS.
- J. A. Gerlt and P. C. Rabbitt, Annu. Rev. Biochem., 2001, 70, 209 CrossRef CAS.
- A. M. Gulick, D. M. Z. Schmidt, J. A. Gerlt and I. Rayment, Biochemistry, 2001, 40, 15716 CrossRef CAS.
- W. S. Yew, E. L. Wise, I. Rayment and J. A. Gerlt, Biochemistry, 2004, 43, 6427 CrossRef CAS.
- E. L. Wise, W. S. Yew, J. A. Gerlt and I. Rayment, Biochemistry, 2004, 43, 6438 CrossRef CAS.
- J. A. Gerlt, P. C. Babbitt and I. Rayment, Arch. Biochem. Biophys., 2005, 433, 59 CrossRef CAS and the references cited therein.
- K. Miyamoto and H. Ohta, J. Am. Chem. Soc., 1990, 112, 4077 CrossRef CAS.
- H. Ohta, Bull. Chem. Soc. Jpn., 1997, 70, 2895 CAS.
- K. Miyamoto and H. Ohta, Biocatalysis, 1991, 5, 49 Search PubMed.
- K. Miyamoto and H. Ohta, Appl. Microbiol. Biotechnol., 1992, 38, 234 CrossRef CAS.
- K. Miyamoto and H. Ohta, Eur. J. Biochem., 1992, 210, 475 CAS.
- K. Matoishi, M. Ueda, K. Miyamoto and H. Ohta, J. Mol. Catal. B: Enzym., 2004, 27, 161 CrossRef CAS.
- Y. Terao, Y. Ijima, H. Kakidani and H. Ohta, Bull. Chem. Soc. Jpn., 2003, 76, 2395 CrossRef CAS.
- K. A. Gallo and J. R. Knowles, Biochemistry, 1993, 32, 3981 CrossRef CAS.
- M. Yohda, H. Okada and H. Kumagai, Biochim. Biophys. Acta, 1991, 1089, 234 CAS.
- K. Watabe, T. Ishikawa, Y. Mukohara and H. Nakamura, J. Bacteriol., 1992, 174, 3461.
- K. Hatakeyama, Y. Asai, Y. Uchida, M. Kobayashi, M. Terasawa and H. Yukawa, Biochem. Biophys. Res. Commun., 1997, 239, 74 CrossRef CAS.
- K. A. Gallo, M. E. Tanner and J. R. Knowles, Biochemistry, 1993, 32, 3991 CrossRef CAS.
- S. Glavas and M. E. Tanner, Biochemistry, 1999, 38, 4106 CrossRef CAS.
- S. Glavas and M. E. Tanner, Biochemistry, 2001, 40, 6199 CrossRef CAS.
- Y. Ijima, K. Matoishi, Y. Terao, N. Doi, H. Yanagawa and H. Ohta, Chem. Commun., 2005, 877 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: 10.1039/b607211a |
| This journal is © The Royal Society of Chemistry 2006 |