Peptides of aminoxy acids as foldamers
Xiang
Li
and
Dan
Yang
*
Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China
First published on 26th May 2006
Abstract
This Feature Article summarizes our efforts in developing a new family of foldamers from α-, β- and γ-aminoxy acids. From a series of conformational studies, we demonstrate that peptides consisting of aminoxy acids adopt several well-defined secondary structures, such as α N–O turns (which feature an eight-membered-ring hydrogen bond), β N–O turns (a nine-membered-ring hydrogen bond), γ N–O turns (a ten-membered-ring hydrogen bond), 1.88 helices (consecutive homochiral α N–O turns), 7/8 helices (alternating α N–O turns and γ-turns), 1.79 helices (consecutive β N–O turns), reverse turns (consecutive heterochiral α N–O turns) and sheet-like structures.
Xiang Li was born in Kunming, China, in 1981. He received his BSc in Chemistry at Fudan University in 2003. Since September 2003, he has been pursuing his PhD with Professor Dan Yang at The University of Hong Kong. He is now exploring the potential applications of α-aminoxy acids in anion recognition and transport. |
Dan Yang was born in Sichuan, China. She received her BSc in Chemistry from Fudan University, P. R. China, in 1985. Through the US–China Chemistry Graduate Program, she obtained an MA in 1988 at Columbia with Professor Ronald Breslow and a PhD from Princeton under the guidance of Professor Daniel Kahne in 1991. She then spent two years as a postdoctoral fellow with Professor Stuart Schreiber at Harvard. In 1993, she joined The University of Hong Kong, where she is currently a Chair Professor of chemistry. Her research interests include asymmetric catalysis and total synthesis, the design and synthesis of novel foldamers and the chemical biology of natural products. |
Introduction
The relationship between the structures and functions of proteins is remarkable. In biological systems, most of the interesting functions performed by proteins, such as molecular recognition, electron transfer and catalysis, are related to unique secondary and tertiary structures. Studies of the structure–activity relationships (SAR) on small molecules interaction with proteins have been instrumental to drug development.In recent years, studies of protein structures and their functions have progressed rapidly so that the mechanisms of many biological processes are now known. Chemists are presently in a position to purposely design unnatural polymers that fold into well-defined secondary structures that may perform desired functions. Gellman1 used the term “foldamers” to describe polymers that have a strong tendency to adopt specific, compact conformations. Several types of foldamers have been developed in the past few years, including peptidomimetic foldamers, single-stranded abiotic foldamers, nucleotidomimetic foldamers and multistranded abiotic foldamers.2
Naturally occurring peptides consisting of fewer than 10 amino acid residues usually do not possess well-defined secondary structures; in addition, they are susceptible to protease degradation. Peptidomimetic foldamers have been developed to achieve better biostablities.1–5 As the most important models in peptidomimetic foldamer chemistry, β-peptides have been investigated and applied widely in biomolecular design.5 Unlike α-peptides, short β-peptides—even those with as few as six residues—can fold into well-defined secondary structures, such as helices, sheets and turns. Because β-peptides have excellent stability toward proteases,6–8 they are widely used as backbone-modified amino acids in drug design.
α-Aminoxy acids are analogs of β-amino acid in which the β-carbon atom is replaced by an oxygen atom. Because of repulsion between the lone pairs of electrons of the nitrogen and oxygen atoms, the backbone of an α-aminoxy acid is more rigid than that of β-amino acid.9 The aminoxy amide bond is resistant to enzymatic degradation; therefore, α-aminoxy acids have been explored as peptidomimetics in several studies.10 We have investigated the novel secondary structures, including turns and helices, that are adopted by α-aminoxy acids. In an endeavor to expand the family of aminoxy acids, we have also found that β- and γ-aminoxy acids, which are analogs of γ- and δ-amino acids, respectively, can form several well-defined secondary structures. In the following sections, we provide brief descriptions of these types of aminoxy acids, their peptides, and their potential behavior as new class of foldamers.

New building blocks: monomers of aminoxy acids
The α N–O turn
The specific biological functions of a biomolecule are, in general, closely related to its stable secondary structure; thus, understanding the secondary structures of designed peptidomimetics is also of great theoretical and pharmaceutical significance, and such studies of α-aminoxy acids are certainly no exception. We have found that when an α-aminoxy acid is incorporated into a peptide backbone, it induces a strong eight-membered-ring intramolecular hydrogen bond between adjacent residues (the so-called α N–O turn).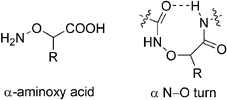
Chiral α-aminoxy acids can be readily prepared from α-amino acids in several steps. Testa et al.11 first reported a systematic method for synthesizing chiral α-aminoxy acids, but the optical purities of the α-aminoxy acids were not reported in literature.10b In this method, the conversion of α-amino acids to α-bromoacids proceeded with mainly retention at the α-carbon atom, whereas nucleophilic displacement of bromide generally followed an SN2 mechanism with an inversion of configuration to give α-aminoxy acids. We used this method to synthesize several D-α-aminoxy acids from natural L-amino acids with overall yields of α-aminoxy esters about 36–55% and the optical purities in the range of 92–94% determined by HPLC analysis.12
We have reported a general method12,13 for the synthesis of doubly protected chiral α-aminoxy acid monomers (Scheme 1). The key step was a Mitsunobu reaction between N-hydroxyphthalimide and α-hydroxy tert-butyl ester, which introduced the N–O segment of α-aminoxy acids and afforded the protected α-aminoxy acids with an inversion of configuration at the α-carbon. The overall yields for syntheses of α-aminoxy acids were in the range of 36–56% and no purification was needed for most of the steps. The optical purities of chiral α-aminoxy acids were found to be 95–99% by HPLC analysis, which indicates that very little or no racemization occurred in the synthesis of chiral α-aminoxy acids. Furthermore, the N-terminal and C-terminal protecting groups can be easily removed under basic and acidic conditions, respectively. Shin et al.14 have synthesized a series of optically active phthaloyl aminoxy acids with nonpolar and polar side chains as building blocks for the preparation of diverse α-aminoxy peptides from α-amino acids and α-hydroxy acids. The C-terminal protecting group, in contrast to tert-butyl in our method, is replaced by benzyl.
 | ||
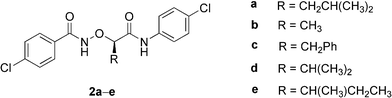
| Scheme 1 Reagents and conditions: i, NaNO2, H2SO4, H2O, 80–95%; ii, AcCl, reflux, 95%; iii, DCC, t-BuOH, DMAP, CH2Cl2, 75–90%; iv, K2CO3, MeOH, H2O, 85–98%; v, N-hydroxyphthalimide, DIAD, PPh3, THF, 69–83%. R = CH2CH(CH3)2, CH3, CH2Ph, CH(CH3)2, CH(CH3)CH2CH3. | ||
The detailed conformational features of oligomers containing α-aminoxy acids were originally investigated within our group.15 Similar to the β-amino acid oligomers, in theory there are several possible intramolecular hydrogen bonds formed within an α-aminoxy acid oligomer. Theoretical calculations on diamide 1 suggest that its most favorable conformation is the rigid eight-membered-ring hydrogen-bonded structure 1a (Fig. 1). The hydrogen bond in 1a is strong, as indicated by the short O⋯H distance and the near linearity of the O⋯H–N bond angle. This calculated conformation was confirmed through X-ray crystallography to occur in the solid-state structure of diamide 2a, which displays a right-handed N–O turn having an ∠NOCαCo dihedral angle of +78.4° (Fig. 2).13
![Structures of α-aminoxy amide 1. The values of Grel (kcal mol−1) were calculated using the HF/6-31G* (MP2/6-31G*) and [HF/6-31G* CHCl3 solvation] models. Reprinted with permission from reference 15. Copyright 1996 American Chemical Society.](/image/article/2006/CC/b602230h/b602230h-f1.gif) | ||
| Fig. 1 Structures of α-aminoxy amide 1. The values of Grel (kcal mol−1) were calculated using the HF/6-31G* (MP2/6-31G*) and [HF/6-31G* CHCl3 solvation] models. Reprinted with permission from reference 15. Copyright 1996 American Chemical Society. | ||
 | ||
| Fig. 2 Solid-state structure of 2a and a summary of the NOEs observed for 2a (5 mM in CDCl3); s, strong NOE; w, weak NOE. | ||
The conformations of α-aminoxy diamides 2a–e in nonpolar solvents were firmly established using a combination of experimental techniques.12,13
FTIR Spectra of the diamides were recorded at very low concentrations so that intermolecular hydrogen bonds were unlikely to form. These IR spectra displayed absorptions for both free (non-hydrogen-bonded) and hydrogen-bonded aminoxy amide N–H stretches, indicating that these compounds exist predominantly in the intramolecular hydrogen-bonded conformations. In our 1H NMR spectroscopic studies of the α-aminoxy diamides, we used two methods to characterize the intramolecular hydrogen bonds. The first was 1H NMR spectroscopic dilution, to determine the concentration dependence of the chemical shifts of the amide protons.16 The second was titration with DMSO-d6, i.e., the gradual addition of a strong hydrogen-bond acceptor to a dilute solution of the α-aminoxy diamide in non-hydrogen-bonding solvent (e.g., CDCl3).17 Our 1H NMR spectroscopic chemical shift data for 2a–e suggest that the N-terminal protons of the diamides are solvent-accessible, whereas the C-terminal ones are hydrogen bonded intramolecularly. The two-dimensional rotating-frame Overhauser effect (2D-ROESY) spectrum of 2a in Fig. 2 suggests that a strong nuclear Overhauser effect (NOE) exists between the NHa and CαH groups, but only a weak NOE between the NHb and CαH groups, indicating that this compound adopts a folded structure that agrees well with its crystal structure (Fig. 2).
We have used the circular dichroism (CD) exciton coupling method18 to determine the handedness of the chiral α N–O turns. The CD spectra of diamides 2a–e (D-configuration), which feature different side chains, displayed strong positive exciton coupling with nearly the same maxima and minima (Fig. 3), indicating that, in the D-configuration, our designed diamides all adopt right-handed α N–O turn structures, irrespective of the nature of their side chains.
 | ||
| Fig. 3 (a) Positive chirality corresponds to the positive dihedral angle ∠NOCαCO in the right-handed turn. (b) CD spectra of diamides 2a–e (0.75 mM) in CH2Cl2. | ||
The formation of the α N–O turn was independent of side chain variation; the orientation of the α N–O turn was determined solely by the configuration of the α-carbon atom. Thus, by changing the chirality of the α-carbon atom from the D configuration to the L configuration, the α N–O turn can be switched from a right-handed to a left-handed structure. Fig. 4 shows the solid-state structures of a pair of α N–O turns, in which their backbones are mirror image of each other. The side-chain-independence of these α N–O turn structures indicates that these systems are subjected to backbone-based control. These results allow us to construct well-defined secondary structures using a variety of side chains.
 | ||
| Fig. 4 Solid-state structures of the right-handed α N–O turn and the left-handed α N–O turn. | ||
The β N–O turn
In our search for novel foldamers, we investigated the behavior of β-aminoxy acids, i.e., structures in which an oxygen atom has replaced the γ-carbon atom of γ-amino acids. Relative to the α-aminoxy acids, the β-aminoxy acids possess an extra carbon atom in their backbone and, thus, provide a greater variety of substitution patterns for their peptides and many opportunities to modulate their hydrogen bonding. Similar to β-amino acids,6,19 β-aminoxy acids can be divided into several subclasses according to their backbone substitution patterns. From our studies of diamides of β2,2-, β3- and β2,3-aminoxy acids, we found that, despite their greater flexibility when compared with those of α-aminoxy acids, most of these diamides adopt a rigid β N–O turn, featuring a nine-membered-ring hydrogen bond between adjacent residues, when incorporated into peptide backbones.
β2,2-Aminoxy acid20 was the first member in the family of β-aminoxy acids that we studied. The crystal structure of diamide 3 revealed a nine-membered-ring hydrogen bond between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Oi and NHi+2 groups; the structure was stabilized further by another six-membered-ring hydrogen bond between the NHi+2 and NOi+1 groups. The N–O bond was positioned anti to the Cα–Cβ bond with an ∠NOCβCα dihedral angle of 172° (Fig. 5(a)). In the solid state, four diamide 3 molecules formed a cyclic ring with four intermolecular hydrogen bonds between them. The C-terminal free carbonyl and N-terminal free amide NH units of one molecule of 3 were hydrogen bonded to two adjacent molecules. This molecules' ability to form hydrogen bonds is fully utilized (Fig. 5(b)).
Oi and NHi+2 groups; the structure was stabilized further by another six-membered-ring hydrogen bond between the NHi+2 and NOi+1 groups. The N–O bond was positioned anti to the Cα–Cβ bond with an ∠NOCβCα dihedral angle of 172° (Fig. 5(a)). In the solid state, four diamide 3 molecules formed a cyclic ring with four intermolecular hydrogen bonds between them. The C-terminal free carbonyl and N-terminal free amide NH units of one molecule of 3 were hydrogen bonded to two adjacent molecules. This molecules' ability to form hydrogen bonds is fully utilized (Fig. 5(b)).

 | ||
| Fig. 5 (a) Solid-state structure and (b) solid-state packing pattern of diamide 3. Part (a) reprinted with permission from reference 20. Copyright 2002 American Chemical Society. | ||
We used 2D NMR spectroscopy studies to confirm that the β N–O turn structure of diamide 3 exists in CDCl3 also. The NOE pattern (medium NOEs between the NHi and CβHi protons, but strong NOEs between the NHi+1 and CβHi protons) observed in the 2D NOESY spectrum of 3 matched that predicted from the X-ray crystallographic structure.
Diamides of β3-aminoxy acids can adopt two different types of β N–O turns depending on the sizes of their side chains.21 The expected β N–O turns involving a nine-membered-ring hydrogen bond between the COi and NHi+2 units, which are stabilized further by another six-membered-ring hydrogen bond between the NHi+2 and NOi+1 units, are observed in the crystal structures of both diamides 4 and 5 (Fig. 6). There are, however, significant differences between these β N–O turns. In diamide 4 (Fig. 6(a)), the N–O bond of the β N–O turn is positioned anti to the Cα–Cβ bond, having a dihedral angle θ of 167.2°; the H⋯H distance between the NHa and CβH units is 3.11 Å, which is longer than that between the NHb and CβH units (2.34 Å). The torsional angles of 4 are comparable to those of the β2,2-aminoxy peptide 3 (Fig. 5), but are distinctly different from those of 5, in which the N–O bond is gauche to the Cα–Cβ bond with a dihedral angle θ of 70.4°. The H⋯H distance between the NHa and CβH units in the solid-state structure of 5 (ca. 2.7 Å) is shorter than that between the NHb and CβH units (ca. 3.9 Å; Fig. 6(b)). 2D NOESY studies of diamides 4 and 5 revealed conformations in nonpolar solvents that are consistent with those observed in the solid state. The NOE pattern observed for 4 is distinctly different from that of 5 (Fig. 6). In particular, the NOE between protons Hb and Hβ of 4 was stronger than that between Ha and Hβ (Fig. 6(c)). In contrast, for 5 we observed a slightly stronger NOE between Ha and Hβ than that between Hb and Hβ (Fig. 6(d)). We were interested in further understanding the specific residue-based changes that bring about this conformational switch. Theoretical calculations on a series of model diamides of β3-aminoxy acids rationalized our experimental results very well. In general, a bulky substituent destabilizes the anti conformation because of greater crowding of the substituents.
 | ||
| Fig. 6 Solid-state structures of diamides (a) 4 and (b) 5 and a summary of the NOEs observed in (c) 4 and (d) 5 in CDCl3 or CD2Cl2; s, strong NOE; w, weak NOE. Reprinted with permission from reference 21(a). Copyright 2004 American Chemical Society. | ||
Gellman et al. used cyclic ring-constrained β-amino acids, which are conformationally rigidified residues exhibiting constrained rotation about their Cα–Cβ bonds, to construct β-peptides having well-defined conformations.22–34 They found that the ring size of the side chain of a β-peptide has a significant effect on its secondary structures. The β-peptides of cyclohexane-containing amino acids prefer to form 14-helix structures, while those of cyclopentane-containing amino acids favor 12-helix formation; these conformations result from the different torsional preferences of the Cα–Cβ bonds in the rings of the individual residues.23,35 To investigate whether this “ring-size effect” applies in our β-aminoxy acids, we explored the conformational features of the β2,3-cyclic aminoxy acids, a subclass of β-aminoxy acids in which the α- and β-carbon atoms comprise part of an aliphatic ring (either a cyclopentane or cyclohexane unit).36Fig. 7 indicates that the most stable conformations of 6 and 7 that we obtained through theoretical calculations were the β N–O turn structures. To maintain the intramolecular hydrogen bonds of the β N–O turns, the torsional angle θ must be adjusted to compensate for changes in the torsional angle ϕ caused by the different ring sizes. The 2D NOESY spectra exhibit very similar NOE patterns for diamides 8 and 9: weak NOEs exist between the NHi and CβHi protons, but strong NOEs exist between the NHi+1 and CβHi protons (Fig. 8). Moreover, these NOE patterns agree well with the corresponding distances between protons in the β N–O turns of the calculated structures of 6 and 7. This correlation suggests that, although the ring size of the side chain varies from five to six atoms, diamides 8 and 9 adopt similar β N–O turn structures in solution, in contrast to Gellman's finding for β-peptides. Furthermore, the solid-state structure of 9, obtained through X-ray crystallography, revealed a β N–O turn structure in which the N–O bond is anti to the Cα–Cβ bond with an ∠NOCαCβ dihedral angle of 172° (Fig. 9). This structure is similar to the β N–O turns observed in the diamides of β2,2-aminoxy acid.
 | ||
| Fig. 7 Lowest energy conformations in the calculated structures of 6 and 7. Reproduced from reference 36 with permission of Wiley. | ||
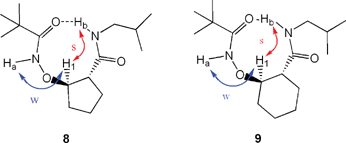 | ||
| Fig. 8 Summary of NOEs observed for compounds 8 and 9 in CDCl3 (s, strong NOE; w, weak NOE). | ||
 | ||
| Fig. 9 Solid-state structure of diamide 9. | ||
The γ N–O turn
To expand the family of aminoxy acid residues and to test the ability of other aminoxy acids to form local intramolecular hydrogen bonds, we have also begun to explore the conformational properties of γ-aminoxy acids,37 which belong to the δ-peptide family. β-Turns of tetrapeptide fragments are common structural features of proteins and, thus, it is not surprising that much activity in the δ-peptide family has been aimed at creating β-turn mimics.2,38–41 To restrict the more flexible backbone of γ-aminoxy acids, initially we examined the effect of incorporating a γ-phenyl substituent. X-Ray crystallographic structural analysis of diamide 10 revealed a β-turn mimic: a γ N–O turn, featuring an intramolecular 10-membered-ring hydrogen bond between the COi and NHi+2 units (Fig. 10(a)). The hydrogen bond distance (O⋯H) is 2.07 Å and the Cγ–O bond is positioned gauche to the Cα–Cβ bond with a ∠CαCβCγO dihedral angle of 69°. In the 2D NOESY spectrum of 10 in CDCl3, the NOE signal between regular amide N–H and γ-H protons indicates a folded backbone consistent with that observed in the solid-state structure of 10 (Fig. 10(b)). 1H NMR spectroscopic data also supports the existence of an intramolecular hydrogen bond between the N–H and CO groups on the two ends of the molecule, suggesting a preference for this secondary structure in solution.
 | ||
| Fig. 10 (a) Solid-state structure of diamide 10. (b) Summary of NOEs observed for diamide 10 in CDCl3. Reprinted with permission from reference 37. Copyright 2004 American Chemical Society. | ||
New foldamers: oligomers of aminoxy acids
As we have discussed above, the monomers of α-, β- and γ-aminoxy acids exhibit several kinds of rigid conformations (the α, β and γ N–O turns, respectively). We were interested in utilizing these novel building blocks to construct peptides having more-diverse secondary structures. Given our focus on oligomers of aminoxy acids, in the following discussion we focus on the structural features of oligomers of aminoxy acids.Helices
α-Aminoxy acids can induce α N–O turn in diamides. The chiral α N–O turn structure is determined by the chirality of the α-carbon atom and is independent of the nature of the side chain. We reasoned that for homochiral oligomers of α-aminoxy acids, eight-membered-ring intramolecular hydrogen bonds should exist between adjacent residues and that these consecutive homochiral α N–O turns should lead to helical structures. Quantum mechanics calculation of the oligomers of homo-D-α-aminoxy acids were performed by Wu et al.42 The calculated structures of tetramer 11 reveal that its lowest-energy conformation both in the gas phase and in solution contain four consecutive right-handed α N–O turns, i.e., it has a helical structure (Fig. 11). The calculated structures reveal several unique features. (1) The backbones of the oligomers of homo-D-α-aminoxy acids form right-handed helical structures, with consecutive eight-membered-ring hydrogen bonds (α N–O turns). The intramolecular hydrogen bonds are lined up along the helical axis with O⋯H–N angles of ca. 157°. (2) The side chains of the oligomers alternate on opposite sides of the helix with a distance of 6.5 Å between the groups at the i and i + 2 positions, a pattern that is reminiscent of the twisted parallel β-sheets found in proteins. (3) The amide carbonyl group at the i + 2 position is twisted by +50° from the one at the i position; this arrangement suggests a 1.88 helix or a twisted 28 helix with two residues per helical turn. | ||
| Fig. 11 Calculated structure of 11. Reprinted with permission from reference 13. Copyright 1999 American Chemical Society. | ||
We performed conformational studies on the oligomers of homo-D-α-aminoxy acids in nonpolar solvents using NMR, IR and CD spectroscopic methods.12,13 The IR and 1H NMR spectra suggest that in each oligomer, except for the N-terminal one, the amide NH group at the i + 2 position is hydrogen bonded intramolecularly to the carbonyl oxygen atom at the i position. In agreement with the results of the theoretical calculation, we observed strong NOEs between the NHi and CαHi protons, but weak NOEs between the NHi+1 and CαHi protons; this pattern is similar to that of the diamides (Fig. 2). The helical conformation was further confirmed through the X-ray crystallographic analysis of triamide 12 (Fig. 12).43 Helix formation in biopolymers is generally length-dependent, with a significant degree of helix formation occurring only after a critical chain length has been reached.5 In organic solvents, natural α-peptides require ca. 10–12 residues to form stable helices and at least six residues are needed for β-peptides. In contrast, we observed helical structures in oligomers of α-aminoxy acids as short as dimers; such structures represent the shortest helices ever found. This discovery opens up an opportunity to design relatively short peptides, which have functionalized side chains, to simulate natural α-peptides interaction with proteins.
 | ||
| Fig. 12 Solid-state structure of diamide 12. | ||
Fig. 13 indicates that the CD curve of monomer 13a is featureless because it did not possess a defined secondary structure. Dipeptide 13b exhibited a slightly weak CD absorption because it only contains one N–O turn. The CD curves of the oligomers 13c–e were almost superimposable—a maximum at 195 nm, a minimum at 225 nm, and a zero crossing in the range of 212–222 nm—indicating the absence of a significant positive cooperative effect. It seems that once the length satisfies the smallest helix, as in the case of tripeptide 13c, elongation of the peptide does not provide extra stability to the helical conformation. The similar CD absorptions of tetramers 13d and 13f, which possess different side chains, suggest that the helix formation is independent of the nature of the side chain; i.e., helix formation from peptides of α-aminoxy acids is controlled by the nature of the backbone.
 | ||
| Fig. 13 CD spectra of compounds 13a–f in 2,2,2-trifluoroethanol. | ||
γ-Turns, which are reversed-turn secondary structures found in proteins, are formed by a 3 → 1 hydrogen bond between the CO group of amino acid residue i and the NH group of amino acid residue i
+ 2.44–46 Although γ-turns are observed less frequently than β-turns in proteins,46 they play important roles in biological recognitions.47–50 It is challenging to investigate the roles of γ-turns in protein–peptide recognition, however, because they seldom exist in short, linear peptides. In an exploration of the conformations of hybrid peptides containing α-amino acids and α-aminoxy acids, we demonstrated that in peptides of alternating D-α-amino acids and L-α-aminoxy acids, the seven-membered-ring intramolecular hydrogen bond, i.e., γ-turn, is initiated by a following α N–O turn. Thus, a novel 7/8 helical structure is induced in this type of peptide.51

Conformational studies, performed using NMR, FT-IR and CD spectroscopic techniques, suggested that oligomers 14b, 14d and 14e form N–O turns and γ-turns simultaneously in solution—even in a protic solvent, methanol. Fig. 14(a) displays the CD spectra of compounds 14a–e in trifluoroethanol after normalizing for the concentration and the number of aminoxy acid residues. Peptide 14c exhibited a CD curve that is unlike those of the other peptides, possibly because the γ-turn between the C-terminal NH unit and the CO group of the aminoxy acid residue of 14c was not formed, as revealed by 1H NMR spectroscopy. The CD curves of oligomers 14d and 14e were almost superimposable—a maximum at 227 nm, a minimum at 195 nm, and a zero crossing at 211 nm—suggesting that oligomers 14d and 14e adopt the same type of secondary structure: a novel mixed 7/8 helix. The CD curves of peptides 14a–e in methanol (Fig. 14(b)) display similar patterns and intensities as those recorded in 2,2,2-trifluoroethanol, suggesting that the mixed 7/8 helix remains unchanged in methanol.
 | ||
| Fig. 14 CD spectra of compounds 14a–e recorded at 25 °C in (a) 2,2,2-trifluoroethanol and (b) methanol. | ||
The structure of this novel 7/8 helical conformation was further confirmed from theoretical calculations. For example, the most stable conformation of tripeptide 15 features an alternating α N–O turn and γ-turn structure (Fig. 15). In the alanine residue, the α-methyl group occupies an equatorial position, which is characteristic of an inverse γ-turn. The Cα–Cβ bonds of the aminoxy acid residues are aligned anti to the N–O bonds.
 | ||
| Fig. 15 Calculated most-stable conformation of tripeptide 15 in CH2Cl2. Reprinted with permission from reference 51. Copyright 2003 American Chemical Society. | ||
Helical structures are also observed in oligomers of β-aminoxy acids. Fig. 16(a) presents the well-defined helical conformation found in the solid-state structure of dipeptide 16, which comprises β2,2-aminoxy acids.20 The helix was composed from two consecutive nine-membered-ring intramolecular hydrogen bonds, i.e., two β N–O turns. The lengths of the hydrogen bonds between the NHi+2 and OCi units were 1.93 Å for the first β N–O turn and 2.29 Å for the second. The shorter NH⋯OC distance in the former turn reflects the higher acidity of an aminoxy amide NH group relative to that of a common amide NH unit. Similar to the structure of diamide 3, each of the β N–O turns of 16 featured an N–O bond aligned anti to the Cα–Cβ bond; their ∠NOCβCα dihedral angle are similar (170 and 174°). The amide carbonyl group at position i
+ 2 was twisted by +65.9° from that at position i, suggesting a novel 1.79 helix. Similar to the 1.88 helices found in peptides of D-α-aminoxy acids, the side chains were projected laterally from the axis of the helix, but, in contrast, the distance between the α-carbon atoms at the i and i
+ 2 positions of the 1.79 helix was longer (7.1 Å) than that found in the 1.88 helix (6.3 Å). In the solid state, when compared with β2,2-aminoxy diamide 3 (Fig. 5(b)), one molecule of compound 16 formed two intermolecular hydrogen bonds with two adjacent molecules in a linear fashion (Fig. 16(b)). A similar 1.89 helical structure, composed from two consecutive β N–O turns, was also observed in the solid state for a tripeptide 17, which comprises β2,3-cyclic aminoxy acids (Fig. 17).36 In the first N–O turn, the length of the O⋯H hydrogen bond between the COi and NHi+2 units is 1.93 Å, which again reflects the relatively higher acidity of aminoxy amide protons. In the second N–O turn, the O⋯H hydrogen bond distance between the COi+1 and NHi+3 units (2.07 Å) is shorter than that present in 9 (2.20 Å), indicating that hydrogen bonding is enhanced upon increasing the number of N–O turns; i.e., a cooperative effect exists.
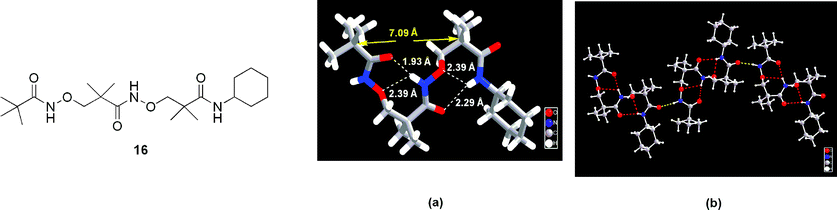 | ||
| Fig. 16 (a) Solid-state structure and (b) solid-state packing pattern of tripeptide 16. Part (a) reprinted with permission from reference 20. Copyright 2002 American Chemical Society. | ||
 | ||
| Fig. 17 Solid-state structure of tripeptide 17. | ||
Because there are two types of β N–O turns induced by β3-aminoxy acids, we used dipeptides 18 and 19,21 which have small (methyl) and large (isobutyl) side chains, respectively, to probe the effect that the side chain has on the helical conformation of a β3-aminoxy peptide. 1H NMR, FT-IR and CD spectroscopy studies of 18 and 19 in nonpolar solvents all suggest that the helical structures possess two nine-membered-ring hydrogen bonds, i.e., two consecutive β N–O turns. Interestingly, we observed two different NOE patterns for dipeptides 18 and 19 (Fig. 18) that are analogous to those observed in the β N–O turns of diamides 4 and 5 (Fig. 6). Therefore, two types of β N–O helices constructed from two types of β N–O turns are dominant in 18 and 19. The N–O bond is aligned anti to the Cα–Cβ bond in 18, which features methyl side chains, but it is gauche in 19, which possesses isobutyl side chains.
 | ||
| Fig. 18 Summary of NOEs observed in dipeptides 18 and 19 in CDCl3 or CD2Cl2 (s, strong NOE; w, weak NOE). | ||
Reverse turns
β-Turns, in which peptide backbones reverse their directions, are common secondary structures of peptides.52–55 We are interested in constructing reverse turns using aminoxy acids as building blocks. We have established that a D-α-aminoxy acid induces a right-handed α N–O turn and its L-enantiomer leads to a left-handed α N–O turn.13,15 While oligomers of homochiral α-aminoxy acids adopt an extended helical structure (1.88 helix),13 we were interested in exploring the conformational features of oligomers of heterochiral α-aminoxy acids. Our theoretical calculations revealed that a loop-like conformation was the most stable structure of dipeptide 20, which consists of two heterochiral α-aminoxy acids, while a helical structure was dominant for dipeptide 21, which contains homochiral α-aminoxy acids (Fig. 19).56 The calculations were confirmed by the solid-state structure of dipeptide 22, in which two heterochiral α N–O turns were characterized by short NH⋯OC distances and ideal N–H⋯O angles (Fig. 20). The conformation of 22 in CDCl3 was determined from 2D NOESY experiments. The NOE pattern of 22 was similar to that observed in the spectrum of two consecutive homochiral α N–O turns; i.e., 22 has a conformational preference for two consecutive α N–O turns in solution.12,13 We used this loop segment to constrain the tetrapeptides 23 and 24 in the form of a reverse-turn structure. 1H NMR spectroscopic dilution and DMSO-d6 addition experiments and 2D-NOESY data indicated that the tetrapeptides 23 and 24 folded into reverse-turn conformations that featured head-to-tail 16-membered-ring intramolecular hydrogen bonds.56
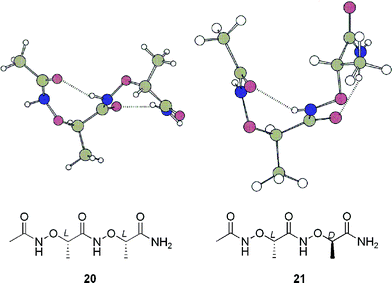 | ||
| Fig. 19 Calculated most-stable conformations of 20 and 21. | ||
 | ||
| Fig. 20 (a) Solid-state structure of dipeptide 22. (b) Summary of NOEs observed in 22 in CDCl3 (s, strong NOE; w, weak NOE). Reprinted in part with permission from reference 56. Copyright 2003 American Chemical Society. | ||

Shin et al. have also reported β-turn mimics prepared using α-aminoxy acids.57 They designed and characterized the α-aminoxy tripeptide 25, which consists of an oxanipecotic acid dimer and an α-aminoxy acid. Conformational studies in solution, performed using FT-IR and NMR spectroscopy, suggested that this compound adopts a loop conformation with consecutive α N–O turn and β turn-like structures. Theoretical calculations of the structure of tripeptide 25 supported these conformational features and agreed well with the NOE data (Fig. 21).
 | ||
| Fig. 21 (a) Energy-minimized structure of 25. (b) Summary of NOEs observed in 25 in CDCl3. Reprinted with permission from reference 57. Copyright 2003 American Chemical Society. | ||
Sheets
In the course of our conformational studies into the oligomers of α-aminoxy acids, we found that the conformation of tripeptide 26 in the solid state was different from that existing in solution. Solution-phase 1H NMR and 2D ROESY studies on tripeptide 26 suggested that it adopts a helical conformation with two consecutive N–O turns. The X-ray crystallographic analysis of tripeptide 26 indicated, however, that no intramolecular hydrogen bond formed; instead, the molecule adopted a more-extended conformation, akin to that of an arched β-strand (Fig. 22).43 The amide bonds lay in the plane of the β-strand and the backbone bulged up and down, directing the side chains above and below the plane of the β-strand. The distance between the side chains at the i and i + 2 positions was 6.34 Å, quite close to the distances found in parallel β-sheet structures of α-peptides. In this special β-strand, all of the amide groups were involved in intermolecular hydrogen bonding in an antiparallel fashion (Fig. 22). Each residue had a gauche NO–CαC(
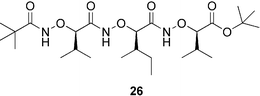 O) torsion angle and the carbonyl groups were oriented in an antiparallel manner, resulting in a sheet with no net dipole. In contrast to the antiparallel β-sheets of α-peptides, the sheet conformation of 26 in the solid state featured consecutive 12- and 16-membered-ring hydrogen bonds. The fact that tripeptide 26 has a helical conformation in solution, but adopts an antiparallel sheet conformation in the solid state, serves as an example to show that the solid-state structure of a molecule does not always reflect its conformation in solution.58
O) torsion angle and the carbonyl groups were oriented in an antiparallel manner, resulting in a sheet with no net dipole. In contrast to the antiparallel β-sheets of α-peptides, the sheet conformation of 26 in the solid state featured consecutive 12- and 16-membered-ring hydrogen bonds. The fact that tripeptide 26 has a helical conformation in solution, but adopts an antiparallel sheet conformation in the solid state, serves as an example to show that the solid-state structure of a molecule does not always reflect its conformation in solution.58
 | ||
| Fig. 22 Antiparallel sheet structure in the packing of tripeptide 26 in the solid state. | ||
Such sheet-like structures were also observed in the solid-state structures of peptides of β3-aminoxy acids.21 The conformation of dipeptide 18 in the solid state led to more-extended parallel sheet-like structures stabilized through intermolecular hydrogen bonds (Fig. 23). In these extended conformations, all of the amide groups are involved in intermolecular 16-membered-ring hydrogen bonding in a parallel fashion. According to the theoretical calculations, the extended conformers of β3-aminoxy acids are less stable than are bent ones (β N–O turns), but the difference in energy between the two is not too great. For dipeptide 18, which has small side chains, it is possible that van der Waals interactions between the protecting groups at both the C- and N-termini result in the sheet structures being more favorable in the solid state, i.e., when the molecules are packed at exceedingly “high concentration.”
 | ||
| Fig. 23 Parallel sheet structure found the packing of dipeptide 18 in the solid state. Reprinted in part with permission from reference 21(a). Copyright 2004 American Chemical Society. | ||
Cyclic peptides
In addition to our studies of linear peptides, we have also prepared and characterized two cyclic hexapeptides containing α-aminoxy acids.59,60Fig. 24 presents the calculated lowest-energy conformation of model compound 27, which comprises DL-α-aminoxy acids. The intramolecularly hydrogen bonded cyclopeptide adopts a bracelet-like conformation with consecutive α N–O turns; the α protons of all of the residues point inward and the side chains decorate two edges of the ring (all of the D-aminoxy acid side chains are located on one side of the bracelet, with all of the L-aminoxy acid side chains on the other).59 This conformation makes 27 quite polar on the inside of the bracelet, but nonpolar on its external surface. Instead of forming consecutive α N–O turns, a similar conformation, featuring alternating α N–O turns and γ turns, was induced in the cyclic hexapeptide 28, which is composed of alternating D-α-aminoxy acids and D-α-amino acids (Fig. 25).60 | ||
| Fig. 24 Calculated lowest-energy conformation of the cyclic hexapeptide 27. Reprinted with permission from reference 59. Copyright 2002 American Chemical Society. | ||
 | ||
| Fig. 25 Calculated lowest-energy conformation of the cyclic hexapeptide 28. Reproduced from reference 60 with permission of Wiley. | ||
Conclusion and outlook
Since we first published—almost ten years ago—our studies of the secondary structures induced by α-aminoxy acids, a surprisingly large number of well-defined secondary structures have been discovered within the family of aminoxy acids. Because of their rigid conformations, aminoxy acids are versatile building blocks with which to construct peptidomimetic foldamers. Peptidomimetics that adopt well-defined secondary structures in buffers or in polar solvents to mimic natural peptide–protein interactions are interesting in drug discovery. Our current studies are, therefore, concentrated on design and synthesis of aminoxy peptides with functional side chains and characterization of their secondary structures in polar solvents and buffers. Considering their biological stability towards proteases, aminoxy peptides that adopt well-defined secondary structures—with their functional side chains positioned appropriately—are expected to have the potential to simulate natural α-peptides, i.e., to bind to target receptors and proteins. We believe that further research in this field will lead to potential applications for these systems in the design of pharmaceuticals, materials and molecular devices.Acknowledgements
We thank Professor Y.-D. Wu and his coworkers for conducting the quantum mechanics calculations on the conformations of aminoxy peptides and Professors M.-J. Zhang, G. D. Brown, H.-Z. Sun and K.-H. Sze for advice on the NMR spectroscopy studies. We are very grateful to our coworkers—Fei-Fu Ng, Zhan-Ji Li, Jin Qu, Bing Li, Wei Li, Yu-Hui Zhang, Yu Hao, Dan-Wei Zhang, Ze-Min Dong, Fei Chen and Ke-Sheng Song—for their enthusiasm and diligent work. We thank the University of Hong Kong, Hong Kong Research Grants Council (HKU 7117/97P, HKU 7098/01P and HKU 7367/03M), Fudan University, the National Natural Science Foundation of China (Project No. 20202001) and the Bristol-Myers Squibb Foundation (Unrestricted Grants in Synthetic Organic Chemistry) for providing financial support.Notes and references
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS.
- M. S. Cubberley and B. L. Iverson, Curr. Opin. Chem. Biol., 2001, 5, 650 CrossRef CAS.
- J. A. Patch and A. E. Barron, Curr. Opin. Chem. Biol., 2002, 6, 872 CrossRef CAS.
- (a) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS; (b) D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015 RSC.
- D. Seebach, M. Overhand, F. N. M. Kuhnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913 CrossRef CAS.
- (a) T. Hintermann and D. Seebach, Chimia, 1997, 50, 244; (b) D. Seebach, S. Abele, J. V. Schreiber, B. Martinoni, A. K. Nussbaum, H. Schild, H. Schulz, H. Hennecke, R. Woessner and F. Bitsch, Chimia, 1998, 52, 734 CAS.
- J. Frackenpohl, P. I. Arvidsson, J. V. Schreiber and D. Seebach, ChemBioChem, 2001, 2, 445 CrossRef CAS.
- Y.-D. Wu, D.-P. Wang, K. W. K. Chan and D. Yang, J. Am. Chem. Soc., 1999, 121, 11189 CrossRef CAS.
- (a) I. Schon, L. Kisfaludy, J. Nafradi, L. Varga and V. Varro, Hoppe-Seyler's Z. Physiol. Chem., 1978, Bd. 359, 897; (b) M. Briggs and J. S. Morley, J. Chem. Soc., Perkin Trans. 1, 1979, 2138 RSC.
- E. Testa, B. J. R. Nicolaus, L. Mariani and G. Pagani, Helv. Chim. Acta, 1963, 46, 766 CAS.
- D. Yang, B. Li, F. F. Ng, Y.-L. Yan, J. Qu and Y.-D. Wu, J. Org. Chem., 2001, 66, 7303 CrossRef CAS.
- D. Yang, J. Qu, B. Li, F.-F. Ng, X.-C. Wang, K.-K. Cheung, D.-P. Wang and Y.-D. Wu, J. Am. Chem. Soc., 1999, 121, 589 CrossRef CAS.
- I. Shin, M. R. Lee, J. Lee, M. Jung, W. Lee and J. Yoon, J. Org. Chem., 2000, 65, 7667 CrossRef CAS.
- D. Yang, F.-F. Ng, Z.-J. Li, Y.-D. Wu, K. W. K. Chan and D.-P. Wang, J. Am. Chem. Soc., 1996, 118, 9794 CrossRef CAS.
- T. S. Haque, J. C. Little and S. H. Gellman, J. Am. Chem. Soc., 1994, 116, 4105 CrossRef CAS.
- G. T. Copeland, E. R. Jarvo and S. J. Miller, J. Org. Chem., 1998, 63, 6784 CrossRef CAS.
- N. Harada and K. Nakanishi, in Circular Dichroic Spectroscopy: Exciton Coupling in Organic Stereochemistry, University Science Books, Mill Valley, CA, 1983 Search PubMed.
- (a) D. Seebach, P. E. Ciceri, M. Overhand, B. Jaun, D. Rigo, L. Oberer, U. Hommel, R. Amstutz and H. Widmer, Helv. Chim. Acta, 1996, 79, 2043 CrossRef CAS; (b) Y.-D. Wu and D.-P. Wang, J. Am. Chem. Soc., 1999, 121, 9352 CrossRef CAS; (c) D. Seebach, S. Abele, K. Gademann, G. Guichard, T. Hintermann, B. Jaun, J. L. Matthews and J. Schreiber, Helv. Chim. Acta, 1998, 81, 932 CrossRef CAS; (d) D. Seebach, K. Gademann, J. L. Matthews, T. Hintermann, B. Jaun, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1997, 80, 2033 CAS.
- D. Yang, Y.-H. Zhang and N.-Y. Zhu, J. Am. Chem. Soc., 2002, 124, 9966 CrossRef CAS.
- (a) D. Yang, Y.-H. Zhang, B. Li, D.-W. Zhang, C.-Y. J. Chan, N.-Y. Zhu, S.-W. Luo and Y.-D. Wu, J. Am. Chem. Soc., 2004, 126, 6956 CrossRef CAS; (b) D. Yang, Y.-H. Zhang, B. Li and D.-W. Zhang, J. Org. Chem., 2004, 69, 7577 CrossRef CAS.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 13071 CrossRef CAS.
- D. H. Appella, L. A. Christianson, D. A. Klein, D. R. Powell, X. Huang, J. J. Barchi, Jr. and S. H. Gellman, Nature, 1997, 387, 381 CrossRef CAS.
- D. H. Appella, L. A. Christianson, D. A. Klein, M. R. Richards, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 7574 CrossRef CAS.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 6206 CrossRef CAS.
- J. J. Barchi, Jr., X. Huang, D. H. Appella, L. A. Christianson, S. R. Durell and S. H. Gellman, J. Am. Chem. Soc., 2000, 122, 2711 CrossRef.
- D. H. Appella, J. J. Barchi, Jr., S. R. Durell and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 2309 CrossRef CAS.
- X. Wang, J. F. Espinosa and S. H. Gellman, J. Am. Chem. Soc., 2000, 122, 4821 CrossRef CAS.
- H.-S. Lee, F. A. Syud, X. Wang and S. H. Gellman, J. Am. Chem. Soc., 2001, 123, 7721 CrossRef CAS.
- M. G. Woll, J. D. Fisk, P. R. LePlae and S. H. Gellman, J. Am. Chem. Soc., 2002, 124, 12447 CrossRef CAS.
- P. R. LePlae, J. D. Fisk, E. A. Porter, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2002, 124, 6820 CrossRef CAS.
- T. L. Raguse, E. A. Porter, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2002, 124, 12774 CrossRef CAS.
- T. L. Raguse, J. R. Lai and S. H. Gellman, J. Am. Chem. Soc., 2003, 125, 5592 CrossRef CAS.
- J.-S. Park, H.-S. Lee, J. R. Lai, B. M. Kim and S. H. Gellman, J. Am. Chem. Soc., 2003, 125, 8539 CrossRef CAS.
- Y. D. Wu and D. P. Wang, J. Am. Chem. Soc., 1998, 120, 13485 CrossRef CAS.
- D. Yang, D.-W. Zhang, Y. Hao, Y.-D. Wu, S.-W. Luo and N.-Y. Zhu, Angew. Chem., Int. Ed., 2004, 43, 6719 CrossRef CAS.
- F. Chen, N.-Y. Zhu and D. Yang, J. Am. Chem. Soc., 2004, 126, 15980 CrossRef CAS.
- E. Graf von Roedern and H. Kessler, Angew. Chem., Int. Ed. Engl., 1994, 33, 687 CrossRef.
- E. Graf von Roedern, E. Lohof, G. Hessler, M. Hoffmann and H. Kessler, J. Am. Chem. Soc., 1996, 118, 10156 CrossRef CAS.
- R. R. Gardner, G.-B. Liang and S. H. Gellman, J. Am. Chem. Soc., 1995, 117, 3280 CrossRef CAS.
- R. R. Gardner, G.-B. Liang and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 1806 CrossRef CAS.
- Y.-D. Wu, D.-P. Wang, K. W. K. Chan and D. Yang, J. Am. Chem. Soc., 1999, 121, 11189 CrossRef CAS.
- J. Qu, PhD Thesis, The University of Hong Kong, 2001.
- G. Némethy and M. P. Printz, Macromolecules, 1972, 5, 755 CrossRef CAS.
- C. Toniolo, Crit. Rev. Biochem., 1980, 9, 1 CrossRef CAS.
- G. D. Rose, L. M. Gierasch and J. A. Smith, Adv. Protein Chem., 1985, 37, 1 CAS.
- A. Milon, T. Miyazawa and T. Higashijima, Biochemistry, 1990, 29, 65 CrossRef CAS.
- M. Coles, V. Sowemimo, D. Scanlon, S. L. A. Munro and D. J. Craik, J. Med. Chem., 1993, 36, 2658 CrossRef CAS.
- S. M. Vogen, O. Prakash, L. Kirnarsky, S. D. Sanderson and S. A. Sherman, J. Pept. Res., 1999, 54, 74 CrossRef CAS.
- A. M. Andrianov and Y. A. Sokolov, J. Biomol. Struct. Dyn., 2003, 20, 603 Search PubMed.
- D. Yang, W. Li, J. Qu, S.-W. Luo and Y.-D. Wu, J. Am. Chem. Soc., 2003, 125, 13018 CrossRef CAS.
- C. M. Venkatachalam, Biopolymers, 1968, 6, 1425 CrossRef CAS.
- P. Y. Chou and G. D. Fasman, J. Mol. Biol., 1977, 115, 135 CrossRef CAS.
- B. L. Sibanda and J. M. Thornton, Nature, 1985, 316, 170 CrossRef CAS.
- C. M. Wilmot and J. M. Thornton, J. Mol. Biol., 1988, 203, 221 CrossRef CAS.
- D. Yang, J. Qu, W. Li, D.-P. Wang, Y. Ren and Y.-D. Wu, J. Am. Chem. Soc., 2003, 125, 14452 CrossRef CAS.
- B. H. Baek, M. R. Lee, K. Y. Kim, U. I. Cho, D. W. Boo and I. Shin, Org. Lett., 2003, 5, 971 CrossRef CAS.
- Y.-J. Chung, B. R. Huck, L. A. Christianson, H. E. Stanger, S. Krauthäuser, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 2000, 122, 3995 CrossRef.
- D. Yang, J. Qu, W. Li, Y.-H. Zhang, Y. Ren, D.-P. Wang and Y.-D. Wu, J. Am. Chem. Soc., 2002, 124, 12410 CrossRef CAS.
- D. Yang, X. Li, Y. Sha and Y.-D. Wu, Chem.–Eur. J., 2005, 11, 3005–3009 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |