In silico evolution of substrate selectivity: comparison of organometallic ruthenium complexes with the anticancer drug cisplatin†‡
Dirk V. Deubel* and Justin Kai-Chi Lau
ETH Zurich, USI Campus, Computational Science, Via Guiseppe Buffi 13, CH-6900, Lugano, Switzerland. E-mail: metals-in-medicine@phys.chem.ethz.ch
First published on 5th May 2006
Abstract
A comparative quantum chemical approach helps to clarify how the selectivity of anticancer metallopharmaceuticals towards potential biological targets can be controlled by metal and ligands.
The success of the anticancer drug cisplatin (1-Cl, cis-[Pt(NH3)2Cl2]) has stimulated the search for new metallopharmaceuticals.2 Various ruthenium complexes have attracted attention.3 Organometallic ruthenium(II) anticancer complexes of the type [Ru(Ar)(en)Cl]+ (6-Cl, Ar = e.g., η6-benzene; en = 1,2-diaminoethane) are of current interest.4 Despite many experimental studies, the discussion of similarities and differences in the anticancer chemistry of cisplatin and Ru(II) complexes has been controversial. In a recent book chapter, the authors summarized ruthenium anticancer complexes in the section entitled: “Ru complexes that mimic cisplatin”,3d whereas others stated that “Ru complexes probably function in a different manner than cisplatin”.3b We believe that this discussion can be rationalized by considering three steps: (i) the binding of metal complexes to biologically relevant functional groups, (ii) the binding of metal complexes to biological macromolecles, and (iii) the effect of metal complexes on entire cells and organisms. While many recent experiments have focused on the biochemistry of the title complexes,4 quantum chemical calculations could provide the best access to transition structures and predict the trends in the reactivity and selectivity of anticancer complexes towards potential biological targets. In view of 80 quantum chemical studies and molecular simulations of platinum anticancer drugs,5 it is surprising that the promising developments of [Ru(Ar)(en)Cl]+ complexes have been ignored by theoreticians, with few recent exceptions.6–8 In one study,6 the authors compare the hydrolysis of arene–en anticancer complexes that contain leaving groups other than chloride. The other study7 includes a comparison of calculated and experimental activation and reaction free energies for the hydrolysis of [Ru(η6-benzene)(en)Cl]+.
We wish to report a combined density functional theory (DFT) and continuum dielectric model (CDM) study at the B3LYP level,8 aiming to compare in a logical manner pharmaceutically relevant reactions of platinum and ruthenium(II) anticancer complexes. As shown in Fig. 1, the archetypal anticancer drug cisplatin (1-Cl) has been mutated successively, obtaining after five generations the organometallic anticancer complex (6-Cl). The chloro complexes are likely activated upon intracellular hydrolysis of metal–chloro bonds, yielding the aqua complexes cis-[Pt(NH3)2(OH2)Cl]+ (1-OH2)–[Ru(Ar)(en)(OH2)]2+ (6-OH2). For each generation, we have investigated the reaction of the aqua complexes with a library of substrates L shown in Fig. 1.10,11Fig. 2 displays the activation free energies (ΔGa) and reaction free energies (ΔGr) for the substitution reactions in eqn (1). The discussion shall not overemphasize the absolute numbers, for the prediction of which we have achieved an accuracy of approximately 4 kcal mol−1.7 We rather focus on the selectivity trends when comparing one generation of metal complexes with the next, starting with 1-OH2 and ending with 6-OH2 (Fig. 1).
| X-OH2 + L → X-L + H2O (X = 1 ⇒ 6) | (1) |
 | ||
| Fig. 1 Metal complexes (top) and library of substrates L (bottom). In parentheses: biological relevance. | ||
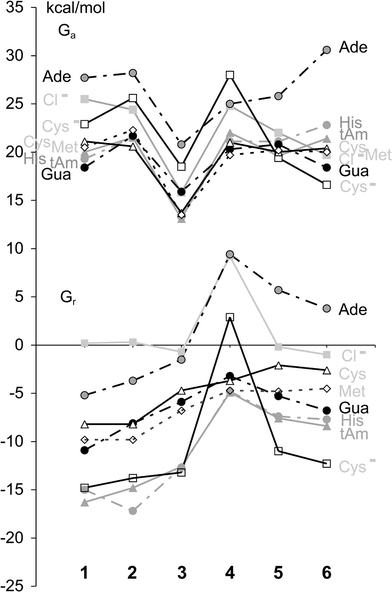 | ||
| Fig. 2 Predicted ΔGa (top) and ΔGr (bottom) of the reactions in eqn (1). | ||
Generation 1 → 2: Replacing the chloro ligand with an ammine ligand causes relatively little changes in ΔGa and ΔGr (Fig. 2).
Generation 2 → 3: Replacing Pt by Pd lowers all activation barriers systematically, indicating an increase in reactivity but no change in kinetic selectivity (Fig. 2). All reactions of the Pd complex (3-OH2), except for the anation, are thermodynamically slightly less favorable than those of the Pt complex (2-OH2). To elucidate these findings, we have performed relativistic and non-relativistic calculations of the reactants, transition states, and products for L = tAm (Fig. 3).12 The calculations confirm the well-known13 trend that the relativistic bond stabilization is stronger for third-row transition metal complexes (Pt) than for second-row transition metal complexes (Pd). The non-relativistic free energy profiles of the Pd and Pt complexes are found to be virtually identical (Fig. 3). Importantly, the calculations show that the relativistic bond stabilization decreases in the order: one strong bond (M–N bond in the products) > one weak bond (M–O bond in the reactants) ≫ two partial bonds (M–O and M–N in the TS). These results explain why the reactions become kinetically much more favorable but thermodynamically slightly less favorable upon replacing Pt by Pd.
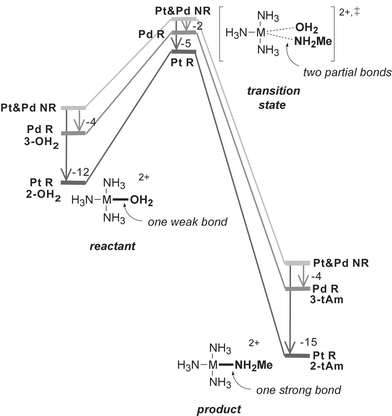 | ||
| Fig. 3 Non-relativistic (NR) and relativistic (R) free energy profiles for the reaction of 2-OH2 (Pt) and 3-OH2 (Pd) with MeNH2 (“tAm”). Arrows: relativistic stabilization of the bond between {M(NH3)3}2+ and OH2 and/or MeNH2 (values in kcal mol−1). | ||
Generation 3 → 4: Moving from {Pd(NH3)3}2+ to {Ru(NH3)5}2+ makes all substitution reactions of the aqua complexes both kinetically and thermodynamically less favorable (Fig. 2). This trend cannot be attributed to steric effects in 4-OH2, because the metalations of the thiol (“Cys”) and the larger thioether (“Met”) residues have similar ΔGa and ΔGr values. The strongest change from 3 to 4 is observed for the thermodynamics of the metalation of the anionic L (Cys− and Cl−). To elucidate this result, we have calculated the amount of charge that is transferred from L to the metal in the transition states and products (for numbers, see ESI‡). The charge transfer from L to the metal increases in the following order: (i) reactions of 4-OH2 < reactions of the other aqua complexes, (ii) reactions of neutral L < reactions of anionic L (Cys− and Cl−), and (iii) transition states < products. It becomes clear that, in the products of anionic L, there is the greatest demand for a charge transfer, but the ability of {Ru(NH3)5}2+ (4) to accept charges is weakest. This result explains why the reactions of 4-OH2 with anionic L become thermodynamically so unfavorable in comparison with those of 3-OH2.
Generation 4 → 5: Creating an organometallic Ru(II) complex by replacing three ammine ligands by one η6-benzene ligand reverses the 3 → 4 trends in ΔGa and ΔGr fully or partially for some of the reactions (Fig. 2). Overall, the trends are (i) fully reversed for the thermodynamics of the reactions with anionic L, (ii) partially reversed for the kinetics of the reactions with anionic L, (iii) partially reversed for the thermodynamics of the reactions with neutral L, and (iv) not reversed for the kinetics of the reactions with neutral L. These results show that the η6-benzene ligand functions as a mediator that adapts its π-acceptor properties to the stereoelectronic requirements in the metal complexes and transition states.
Generation 5 → 6: Replacing the two remaining ammines by the en chelating ligand causes little change in ΔGa and ΔGr for most reactions (Fig. 2). Importantly, Gua metalation with 6-OH2 becomes more favorable, whereas Ade metalation becomes kinetically less favorable. The enhancement of the selectivity to Gua in the en complex can be partially attributed to a conformational activation of the reactant. The transition structure for the reaction of 6-OH2 with Gua shows a strong N–H⋯O6 hydrogen bond (Fig. 4). In the reactant [Ru(Ar)(en)(OH2)]2+ (6-OH2), an N–H bond of the en ligand already points in the direction where Gua–O6 will be located in the TS. In contrast, 5-OH2 is not conformationally activated (see ESI‡). Remarkably, the metal–nucleophile and metal–leaving group distances in the transition structures for all Ru(II) complexes considered herein (4-OH2, 5-OH2 and 6-OH2) are much longer than those in the reactions of the Group 10 complexes. This result is interesting in light of former work, because experimental activation parameters4f,14 suggested ligand substitution reactions of organometallic arene–en Ru(II) complexes to be more associative than those of the inorganic Ru(II) complexes. In the ESI,‡ we have defined a new protocol for analyzing transition structures of ligand-substitution reactions.
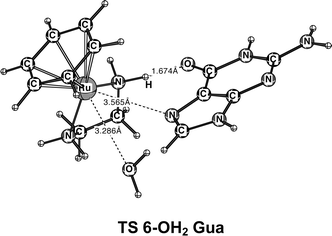 | ||
| Fig. 4 Structure of the transition state for the reaction of 6-OH2 with Gua. | ||
We conclude that, regarding the selectivity towards biologically relevant functional groups (Fig. 2), organometallic Ru(II) anticancer complexes are more similar to cisplatin than to inorganic Ru(II).15 Both cisplatin and en–arene Ru(II) complexes strongly bind to Gua sites of genomic DNA. The latter complexes do not bind to Ade, and they form monofunctional DNA adducts that are recognized and repaired in the cell in a manner different from the bifunctional DNA adducts of cisplatin.4i Nevertheless, the formation and processing of DNA adducts leads in both cases to cell death,4b,j which arises partly from the selectivity of the complexes towards Gua. Given the difference in the chemical structure of cisplatin and organometallic [Ru(Ar)(en)Cl]+ complexes and the striking similarity in their selectivity to biomolecules, we believe that the in silico evolution of substrate selectivity has a promising potential in the virtual screening of new metallopharmaceuticals.
This work has been supported by Prof. M. Parrinello, the Swiss National Science Foundation, the Fonds der Chemischen Industrie, Germany, the Bundesministerium for Bildung and Forschung, Germany, and the Swiss National Computing Center.
Notes and references
- Part V: D. V. Deubel, J. Am. Chem. Soc., 2006, 128, 1654 Search PubMed.
- (a) B. Lippert, Cisplatin; Wiley-VCH, Weinheim, 1999 Search PubMed; (b) Z. J. Guo and P. J. Sadler, Angew. Chem., 1999, 111, 1610 (Angew. Chem., Int. Ed., 1999, 38, 1512) CrossRef; (c) M. A. Fuertes, C. Alonso and J. M. Pérez, Chem. Rev., 2003, 103, 645 CrossRef CAS; (d) J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3611 CrossRef CAS; (e) M. A. Jakupec, M. Galanski and B. K. Keppler, Rev. Physiol. Biochem. Pharmacol., 2003, 146, 1 Search PubMed; (f) D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307 CrossRef CAS.
- (a) M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511 CrossRef CAS; (b) M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209 CrossRef CAS; (c) E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Met. Ions Biol. Syst., 2004, 42, 323 CAS; (d) O. Lentzen, C. Moucheron and A. Kirsch-De Mesmaeker, in Metallotherapeutic Drugs & Metal-Based Diagnostic Agents, ed. M. Gielen and E. R. T. Tiekink, Wiley, Chichester, 2005, p. 359 Search PubMed.
- (a) R. E. Morris, R. E. Aird, P. D. Murdoch, H. M. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616 CrossRef CAS; (b) R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652 CrossRef CAS; (c) H. M. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064 CrossRef CAS; (d) H. M. Chen, J. A. Parkinson, R. E. Morris and P. J. Sadler, J. Am. Chem. Soc., 2003, 125, 173 CrossRef CAS; (e) H. M. Chen, J. A. Parkinson, O. Novakova, J. Bella, F. Y. Wang, A. Dawson, R. Gould, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14623 CrossRef CAS; (f) F. Wang, H. M. Chen, S. Parsons, L. D. H. Oswald, J. E. Davidson and P. J. Sadler, Chem.-Eur. J., 2003, 9, 5810 CrossRef CAS; (g) R. Fernandez, M. Melchart, A. Habtemariam, S. Parsons and P. J. Sadler, Chem.-Eur. J., 2004, 10, 5173 CrossRef CAS; (h) F. Wang, J. Bella, J. A. Parkinson and P. J. Sadler, JBIC, J. Biol. Inorg. Chem., 2005, 10, 147 CrossRef CAS; (i) O. Novakova, J. Kasparkova, V. Bursova, C. Hofr, M. Vojtiskova, H. M. Chen, P. J. Sadler and V. Brabec, Chem. Biol., 2005, 12, 121 CrossRef CAS; (j) Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764 RSC; (k) F. Wang, J. Xu, A. Habtemariam, J. Bella and P. J. Sadler, J. Am. Chem. Soc., 2005, 127, 17734 CrossRef CAS; (l) A. F. A. Peacock, A. Habtemariam, R. Fernandez, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739 CrossRef CAS.
- D. V. Deubel, Chem. Rev. Search PubMed in preparation.
- F. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernandez, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269 CrossRef CAS.
- J. K.-C. Lau and D. V. Deubel, J. Chem. Theory Comput., 2006, 2, 103 Search PubMed.
- For studies on other arene–Ru(II) complexes, see: (a) A. Dorcier, P. J. Dyson, C. Gossens, R. Scopelliti, I. Tavernelli and U. Rothlisberger, Organometallics, 2005, 24, 2114 CrossRef CAS; (b) C. Scolaro, T. J. Geldbach, S. Rochat, A. Dorcier, C. Gossens, A. Bergamo, M. Cocchietto, I. Tavernelli, G. Sava, U. Rothlisberger and P. J. Dyson, Organometallics, 2006, 25, 756 CrossRef CAS.
- For computational details, see ESI‡ or ref. 6.
- See for example: (a) D. V. Deubel, J. Am. Chem. Soc., 2002, 124, 5834 CrossRef CAS; (b) D. V. Deubel, J. Am. Chem. Soc., 2004, 126, 5999 CrossRef CAS; (c) J. K.-C. Lau and D. V. Deubel, Chem.-Eur. J., 2005, 11, 2849 CrossRef CAS.
- The hydrolysis of metal–chloro bonds in 1-Cl–6-Cl is included by studying the anation of the aqua complexes 1-OH2–6-OH2. For simplicity, we have used the three-letter code of amino acids for their functional groups; e.g., “Met” represents dimethylsulfide, the thioether functional group of methionine.
- See for example: A. Diefenbach and F. M. Bickelhaupt, J. Chem. Phys., 2001, 115, 4030 Search PubMed.
- See for example: (a) P. Pyykkö, Angew. Chem., 2002, 114, 3723 (Angew. Chem., Int. Ed., 2002, 41, 3573) CrossRef; (b) P. Schwerdtfeger, Relativistic Electronic Structure Theory, Elsevier, Amsterdam, Part I, 2002; Part 2, 2004 Search PubMed.
- (a) R. van Eldik, Chem. Rev., 2005, 105, 1917 CrossRef CAS; (b) L. Helm and A. E. Merbach, Chem. Rev., 2005, 105, 1923 CrossRef CAS; (c) F. P. Rotzinger, Chem. Rev., 2005, 105, 2003 CrossRef CAS.
- Following the logic of the computational design (Fig. 1), [Ru(NH3)5Cl]+ complexes are herein considered “inorganic Ru(II)”. Note that fac-[Ru(NH3)3Cl3] rather than [Ru(NH3)5Cl]+ is anticancer-active, and two other inorganic Ru complexes, NAMI-A and KP1019 (for structural drawings, see ESI‡), are in clinical trials. Ru(III) complexes are likely reduced in vivo to their Ru(II) analogs;2 future computational studies are required to explore their pharmacologically relevant redox and ligand-substitution reactions.
Footnotes |
| † Quantum chemical studies of metals in medicine. Part VI.1 |
| ‡ Electronic supplementary information (ESI) available: Partial charges, analysis of inorganic reaction mechanisms, selected transition structures, structure of NAMI-A and KP1019, computational details. See DOI: 10.1039/b601590e |
| This journal is © The Royal Society of Chemistry 2006 |