Building three-dimensional nanostructures with active enzymes by surface templated layer-by-layer assembly†
Sakandar
Rauf
a,
Dejian
Zhou
*ab,
Chris
Abell
b,
David
Klenerman
b and
Dae-Joon
Kang
*ac
aNanoscience Centre, University of Cambridge, 11 J J Thomson Avenue, Cambridge, UK CB3 0FF
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: dz209@cam.ac.uk; Tel: (+44) 1223-330107; Fax: (+44) 1223-336362
cSungkyunkwan Advanced Institute of Nanotechnology, Institute of Basic Science, CNNC and Department of Physics, Sungkyunkwan University, Suwon, 440-746, Korea. E-mail: djkang@skku.edu; Tel: (+82) 31-290-5906; Fax: (+82) 31-290-5947
First published on 9th March 2006
Abstract
We show that well-defined three-dimensional nanostructures of functional enzymes can be controllably fabricated by layer-by-layer assembly of avidin and biotinylated horseradish peroxidase on micro-contact printing patterned surface templates.
The controlled positioning of functional biomolecules on surfaces at the micro- to nano scale is important to the development of array-based high-throughput screening techniques, novel biosensors, bioelectronics, tissue engineering, and fundamental studies in cell biology.1 This has been achieved by inkjet printing, micro-fluidics, micro-contact printing (μCP), e-beam lithography, and scanning probe based techniques.2 Most of these arrays however, contained just a single layer of biomolecules.2 This may limit their applications as highly sensitive sensors, since a multilayer structure is often required to increase the sensitivity.3 Layer-by-layer (LBL) assembly4 on patterned surfaces have been shown as an attractive approach to achieve multilayer microscale structures with polymers,5 nanoparticles,6 and biomolecules.7
We are particularly interested in the assembly of well-defined three-dimensional (3D) structures with active enzymes, because they hold important applications in miniaturised assays, sensors, and devices.8 Despite extensive LBL assembly studies with biomolecules, however, to our knowledge, multilayer 3D micro-/nano structures with active enzymes have not been realized.7,9,10 Presumably nonspecific interactions associated with proteins can lead to nonspecific adsorption on the resist background. To obtain well-defined multilayer structures, the nonspecific adsorption must be minimized. Here, we use a self-assembled monolayer (SAM) terminated with hexa(ethylene glycol) groups, known for its ability to reduce nonspecific biomolecule adsorption,11 as our resistive coating. We choose horseradish peroxidase each labelled with on average 2.3 biotins (biotin-HRP) as our model enzyme and avidin as a biocompatible linker because each avidin has 4 biotin binding sites, two on each side of the protein.12 This takes the advantage of the extremely strong biospecific binding between the avidin and biotin (dissociation constant, Kd ∼ 10−15 M).12 HRP labelled with 2.3 biotins is expected to be sufficient since biotinylation occurs mainly on opposite sites on the HRP molecule, preventing the two biotins from the same HRP binding to the same avidin.9
Scheme 1 shows the schematic of our approach (the ideal case). μCP is used to pattern a gold surface with a well-defined SAM template, with regions promoting the assembly of enzymes within a resist background. This is achieved by printing micron-sized SAM stripes of 11-mercaptoundecylhexa(ethylene glycol) alcohol (EG6OH)7 as the resistive coating, followed by filling the unstamped gold regions with a SAM of a biotin terminated thiol, by solution self-assembly.7 Avidin is then specifically assembled onto the biotinylated SAM regions via two of its biotin-binding sites. This leaves two further biotin-binding sites for biotin-HRP binding. This in turn produces a biotin terminated outmost layer for binding of a second avidin layer. Repeating the LBL assembly with avidin and biotin-HRP will produce a controlled growth of the 3D enzyme structures. Since the enzymes are assembled on top of a bio-compatible protein (avidin) layer, the immobilized enzymes should maintain their catalytic function.9 It was anticipated that such 3D enzyme nanostructures (with multiple layers of active enzymes) should exhibit higher overall catalytic activities than conventional immobilized enzyme arrays, which usually contain just a single layer of enzymes.
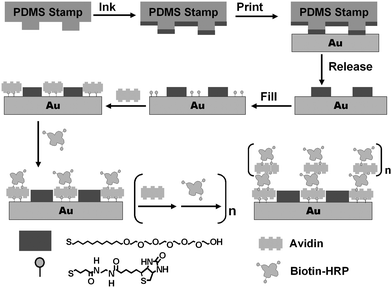 | ||
| Scheme 1 Schematic of our approach to assemble 3D structures with active enzymes. | ||
To confirm that multilayer avidin/biotin-HRP films can be assembled controllably, we assembled homogenous films on gold-coated silicon surfaces (∼200 nm gold with ∼10 nm chromium) and used ellipsometry to monitor the film thickness as a function of the number of assembly cycles.13 The gold surface was coated with a SAM of the biotin-terminated thiol by incubating it in a biotin-disulfide solution.7 The first avidin layer was prepared by incubating the surface with an avidin solution for 30 min. After rinsing with PBS, the surface was incubated with a biotin-HRP solution for 30 min followed by rinsing with PBS. This constituted an assembly cycle (denoted as (avidin/biotin-HRP)1). The surfaces were thoroughly washed but not dried between each assembly step. A specific number of bilayers was made before each measurement. This precaution was found to be critical to maintain the protein function by reducing the possibility of protein denaturation due to drying.
Fig. 1 shows the thickness of the avidin/biotin-HRP film as a function of the number of assembly cycles. It is clear that the protein film thickness increases linearly with the number of the assembly cycles (R = 0.998), suggesting a regular and well-controlled LBL build-up of the enzyme film after the first bilayer. The average thickness of each avidin/biotin-HRP bilayer, ∼1.44 nm is, however, significantly smaller than the combined thickness of closely packed HRP and avidin layers (∼8–10 nm, estimated from the dimensions of the proteins).14 This suggests that the proteins are not closely packed within the film, and similar results have been reported by other groups.10 This is not surprising because the first avidin layer is not densely packed, with a thickness of ∼2.3 nm, which is much smaller than a thickness of ∼5.5 nm expected from a densely packed avidin layer (the size of avidin is ∼4.0 nm × 5.5 nm × 5.5 nm).14
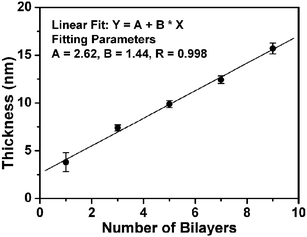 | ||
| Fig. 1 Plot of the avidin/biotin-HRP film thickness vs the number of the assembly bilayers. | ||
Next we assembled the enzymes on the μCP patterned surface templates under identical assembly conditions as those used in the ellipsommetry studies. We used atomic force microscopy (AFM, tapping mode in air) to monitor the topographic evolution of the enzyme patterns following the LBL assembly process.6,7Fig. 2A shows the topographic image of a μCP-patterned SAM template, with 2 µm SAM stripes of the EG6OH separated by the biotin-thiol stripes, on a flat thin-gold coated surface. The EG6OH stripes are ∼2.4 nm taller than the biotin-thiol stripes. This agrees well with the thickness difference between the two SAMs (the EG6OH and biotin-thiol SAMs are found to be ∼3.4 and ∼1.0 nm, respectively).7 After a bilayer of avidin/biotin-HRP is assembled onto the template, the topographic contrast between the protein and EG6OH stripes is reversed (Fig. 2B). The protein stripes are now ∼1.2 nm higher than the EG6OH stripes, presumably because the proteins preferentially assembled onto the biotin stripes. Despite using the EG6OH SAM, one of the most effective resistive coatings to biomolecules, some non-specific adsorption on the resist stripes was still observed.16 We found most of the non-specifically adsorbed proteins could be removed by treating the surface with a surfactant (0.1% Tween 20 in PBS) for 1.5 h. The treatment resulted in an increased height contrast between the two stripes to ∼3.2 nm (Fig. 2C). Further, we found that rinsing with the surfactant after each assembly step significantly reduces nonspecific adsorption on the resist stripes while still maintaining the specific assembly on the functional stripes. Each sample with a given number of bilayers was individually prepared, because the samples need to be thoroughly rinsed with water to remove salt and dried before the AFM measurements. Such treatment may partially denature the assembled proteins.
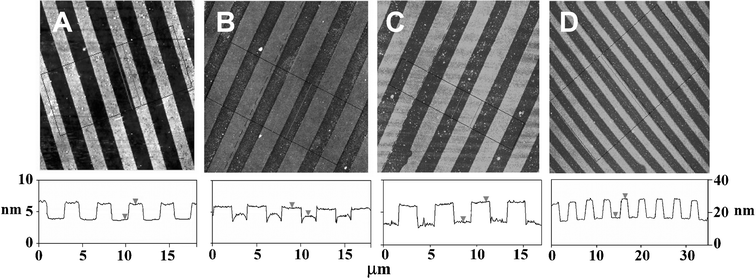 | ||
| Fig. 2 AFM topographic images showing the evolution of the avidin/biotin-HRP 3D structures. Images sizes, 20 µm × 20 µm for A, B, and C, and 40 µm × 40 µm for D. Images A, B, and C are on the left height scale, and image D is on a different scale shown on the right. (A) A μCP-patterned surface, the (EG)6OH stripes are ∼2.4 nm taller than the biotin-thiol stripes. (B) After assembly of a bilayer of avidin/biotin-HRP, the protein stripes are ∼1.2 nm higher than the EG6OH stripes. (C) The same as B after a treatment with the surfactant, the protein stripes are ∼3.0 nm higher than the EG6OH stripes. (D) After assembly of 9 bilayers of avidin/biotin-HRP, the protein stripes are ∼12 nm higher than the EG6OH background. | ||
By using a surfactant rinse in between each assembly step, multi-layered 3D structures of avidin/enzyme can be readily fabricated. Fig. 2D shows the topographic image of the patterned surface after the assembly of 9 avidin/biotin-HRP bilayers. The height of the enzyme strips is now ∼12 nm above the resist background. This corresponds to a total protein layer thickness of ∼14.4 nm, taking into account the original thickness difference between the SAMs. This thickness broadly agrees with that of the ellipsommetry datum (∼15 nm). The topographic image also shows the resist background regions are reasonably clean, with little nonspecific adsorption on the EG6OH region, confirming a well-controlled layer-by-layer build-up of the 3D enzyme structures on the functional pattern region.
An absorption-based assay was used to investigate the activity of the immobilized homogenous enzyme films prepared rinsing with PBS only in between each assembly step without the surfactant. In the presence of H2O2, the HRP catalyses the turnover of a non-coloured substrate, Amplex red, into a coloured product, resorufin, that strongly absorbs at 571 nm.15 The assay was carried out in PBS with 5 mM H2O2 and 25 µM Amplex red. The changes in absorbance at 571 nm at different time intervals for the 1, 5 and 9 bilayer samples were investigated (Fig. S1).† All three samples are catalytically active, and the overall activity increased with the number of the enzyme layers. The enzyme activity in first bilayer is estimated to be ∼14% that of the free enzymes in solution.† This level of catalytic activity is somewhat better than some covalently immobilised HRPs on flat surfaces, where a ∼5% of the free enzyme activity has been reported.17 The rate of activity increase as a function of enzyme layers is sub-linear, with the 5- and 9-bilayer films being only ∼25% and ∼40% more active than the first bilayer. The rate of the catalytic activity increase is slower than those immobilized on submicron beads, where multilayer enzymes produced up to 5-fold increase in the total catalytic activity.9 Presumably the beads have a bigger surface area, and can also diffuse in solution to improve substrate accessibility. Nonetheless, we have achieved an improved overall catalytic activity per unit surface area by using the multilayer enzyme structure.
The fact that the 9-bilayer sample is only 40% more active than the first bilayer suggests that the enzyme activity of the multilayer film mainly comes from the outmost enzyme layer, with a minor contribution from the inner layers. Presumably the exposed outmost enzymes are most directly accessible to the substrates, those at the inner layers have to rely on substrate diffusing through the outer protein layers to reach them, and thus have a much lower substrate accessibility (and hence the apparent activity). The contribution from the inner enzymes is reflected by the slightly higher activity of the 9-bilayer film as compared to the 5-bilayer. The use of substrate flow, rigid spacers (to improve substrate diffusion), and miniaturization of the feature size to the nanoscale (to increase accessibility from side surfaces) should improve the substrate accessibility to these multilayer enzyme structures, and hence the overall enzyme activity.
In summary, we have successfully fabricated well-defined multilayer 3D enzyme structures by using μCP-patterned SAM templates to guide the layer-by-layer assembly of avidin and biotin-HRP. This precisely controls the height and the positions of the enzyme structures assembled on surface. The assembled enzymes maintain some catalytic activity, with the overall activity increasing with the increasing number of the enzyme layers. To our knowledge, this is the first example of multilayer 3D structures assembled from active enzymes. Miniaturization of the structures to the nanoscale should further improve the activity of such enzyme assemblies. Furthermore, operating in 3D would allow more functions, unavailable from 2Ds, to be realised. For example, bi-enzyme and multi-enzyme systems can also be introduced into these 3D structures to achieve higher sensitivity.18 These 3D enzyme nanostructures may have wide applications in highly miniaturized biosensors, biocatalysts, tissue engineering, biochips, and to study biological processes.
We acknowledge the financial support of Interdisciplinary Research Collaboration in Nanotechnology, UK. This work was also supported by the SRC program (Center for Nanotubes and Nanosructured Composites) of MOST/KOSEF and, in part, the Ministry of Science and Technology of Korea through the Cavendish-KAIST Cooperative Research Program.
Notes and references
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Y. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335 Search PubMed.
- T. Okamoto, T. Suzuki and N. Yamamoto, Nat. Biotechnol., 2000, 18, 438 CrossRef CAS; E. Delamarche, A. Bernard, H. Schmid, B. Michel and H. Biebuyck, Science, 1997, 276, 779 CrossRef CAS; K. B. Lee, S. J. Park, C. A. Mirkin, J. C. Smith and M. Mrksich, Science, 2002, 295, 1702 CrossRef CAS; A. Bruckbauer, D. J. Zhou, L. M. Ying, Y. E. Korchev, C. Abell and D. Klenerman, J. Am. Chem. Soc., 2003, 125, 9834 CrossRef CAS; G. J. Leggett, Analyst, 2005, 130, 259 RSC.
- M. K. Beissenhirtz, F. W. Scheller, W. F. M. Stöcklein, D. G. Kurth, H. Möhwald and F. Lisdat, Angew. Chem., Int. Ed., 2004, 43, 4357 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS; P. T. Hammond, Adv. Mater., 2004, 16, 1271 CrossRef CAS.
- X. P. Jiang, S. L. Clark and P. T. Hammond, Adv. Mater., 2001, 13, 1669 CrossRef CAS.
- D. J. Zhou, A. Bruckbauer, C. Abell, D. Klenerman and D.-J. Kang, Adv. Mater., 2005, 17, 1243 CrossRef CAS.
- D. Zhou, A. Bruckbauer, L. Ying, C. Abell and D. Klenerman, Nano Lett., 2003, 3, 1517 CrossRef CAS; A. Biebricher, A. Paul, T. Tinnefeld, A. Golzhauser and M. Sauer, J. Biotechnol., 2004, 112, 97 CrossRef CAS.
- B. Limoges, J. M. Savéant and D. Yazidi, J. Am. Chem. Soc., 2003, 125, 9192 CrossRef CAS.
- S. V. Rao, K. W. Anderson and L. G. Bachas, Biotechnol. Bioeng., 1999, 65, 389 CrossRef CAS.
- X. Cui, R. Pei, X. Wang, F. Yang, Y. Ma, S. Dong and X. Yang, Biosens. Bioelectron., 2003, 18, 59 CrossRef CAS; L. Shi, Y. Lu, J. Zhang, C. Sun, J. Liu and J. Shen, Biomacromolecules, 2003, 4, 1161 CrossRef CAS; L. Shen and N. Hu, Biomacromolecules, 2005, 6, 1475 CrossRef CAS; M. K. Beissenhirtz, F. W. Scheller and F. Lisdat, Anal. Chem., 2004, 76, 4665 CrossRef CAS; H. Ai, M. Fang, S. A. Jones and Y. M. Lovov, Biomacromolecules, 2002, 3, 560 CrossRef CAS.
- K. L. Prime and G. M. Whitesides, Science, 1991, 252, 1164 CrossRef CAS.
- N. M. Green, Adv. Protein Chem., 1975, 29, 85 CAS; O. Livnah, E. A. Bayer, M. Wilchek and J. L. Sussman, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5076 CAS.
- X. Z. Wang, D. J. Zhou, T. Rayment and C. Abell, Chem. Commun., 2003, 474 RSC.
- A. Henriksen, A. T. Smith and M. Gajhede, J. Biol. Chem., 1999, 274, 35005 CrossRef CAS; L. Pugliese, A. Coda, M. Malcovati and M. Bolognesi, J. Mol. Biol., 1993, 231, 698 CrossRef CAS.
- R. Haugland, Handbook of Fluorescent Probes and Research Products, Molecular Probes, Inc., Eugene, Oregon, 9th edn, 2002 Search PubMed.
- D. J. Zhou, X. Z. Wang, L. Birch, T. Rayment and C. Abell, Langmuir, 2003, 19, 10557 CrossRef CAS.
- F. Vianello, L. Zennaro, M. L. Di Paolo, A. Rigo, C. Malacarne and M. Scarpa, Biotechnol. Bioeng., 2000, 68, 488 CrossRef CAS.
- C. M. Niemeyer, J. Koehler and C. Wuerdemann, ChemBioChem, 2002, 3, 242 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental procedures, materials and methods, enzyme activity and AFM measurements.. See DOI: 10.1039/b517557g |
| This journal is © The Royal Society of Chemistry 2006 |