Iridium-complex modified CdSe/ZnS quantum dots; a conceptual design for bifunctionality toward imaging and photosensitization†
Jia-Ming
Hsieh
a,
Mei-Lin
Ho
a,
Pei-Wen
Wu
a,
Pi-Tai
Chou
*a,
Tsai-Tsung
Tsai
b and
Yun
Chi
*b
aDepartment of Chemistry, National Taiwan University, Taipei 106, Taiwan. E-mail: chop@ntu.edu.tw; Fax: +886 (2) 2369 5208; Tel: +886 (2) 2364 3876 ext. 3988
bDepartment of Chemistry, National Tsing Hua University, Hsinchu 300, Taiwan. E-mail: ychi@mx.nthu.edu.tw
First published on 13th January 2006
Abstract
We report the design and synthesis of Ir-complex functionalized CdSe/ZnS quantum dots (QDs), in which the QD plays a key role in imaging, while the Ir-complex acts as a sensitizer to produce singlet oxygen; this conceptual design presents a novel scheme in both bio-imaging and photodynamic therapy.
Ligand-protected nanoparticles consisting of semiconductor cores surrounded by organic monolayers have attracted considerable interest for applications. The interest in these nanomaterials is motivated by the unique optical and electrical properties of the semiconductor cores induced by the quantum dot size effect, while the organic surroundings can provide stability and additional functionality. These unique properties have made them promising nanomaterials for various potential applications including electronics,1 optics,2 and biosensors.3 For biological application, quantum dots possess several characteristics that make them potential photosensitizers,4 and they have recently earned the spotlight as imaging agents5 and diagnostics.6 As for the next stage for the therapeutics, a technique known as photodynamic therapy (PDT)7 has been attracting much attention. Studies have shown that PDT can be as effective as surgery or radiation therapy in treating certain kinds of cancers and precancerous conditions, and may have some advantages: it is less invasive than surgery, it can be targeted very precisely, and it can be repeated several times at the same site if necessary, resulting in less blemishing. PDT involves using a photosensitizing agent (or drug), which is activated by being exposed to light, producing singlet oxygen to destroy cancer cells. In view of exploiting semiconductor nanoparticles in PDT, recent work by Samia5a and co-workers reported that CdSe cores may be used directly to generate singlet oxygen in toluene. However, the yield of singlet oxygen is as low as ∼ 5%, in comparison to ≥ 40% reported for classic photosensitizers.
Herein, we report the conceptual design of a bifunctional system, in which highly luminescent CdSe/ZnS quantum dots (QDs) act as a visible imaging dye, while the third-row transition metal complexes are attached and exploited as a photosensitizer. The size of the CdSe core in CdSe/ZnS QDs can be strategically fine-tuned, so that its emission locates at the low-lying triplet state absorption of the transition metal complexes. Due to the forbidden nature in the triplet manifold, the Förster type of resonance energy transfer can be either drastically reduced or even eliminated, if one can tune the thickness of ZnS and the length of the spacer between QDs and the transition metal complex to a certain long distance. Thus, the luminescent QDs provide the capability for imaging, while the Ir complexes enhanced ultrafast intersystem crossing guarantees unity population at the triplet states, consequently inducing the sensitization of active molecular species, i.e., singlet oxygen, that are toxic to cells and tissues.8,9
Scheme 1 depicts the synthetic route for the Ir–CdSe/ZnS QDs, in which CdSe/ZnS QDs were prepared from CdO using a two-step procedure reported previously.10 A detailed method of preparing hydroxyl substituted pyridyl pyrazole ligand (L1) is elaborated in the supporting information (SI). The first Ir-complex, [(piq)2Ir(L1)], was synthesized via a reaction of [(piq)2IrCl]2 and L1.11 The thio-attached [(piq)2Ir(L1)], i.e. [(piq)2Ir(L2)] (see Scheme 1), was synthesized from a mixture of [(piq)2Ir(L1)] (100 mg, 0.125 mmol), thiotic acid (26 mg, 0.125 mmol), N,N′-dicyclohexylcarbodiimide (52 mg, 0.262 mmol) and N,N′-dimethylamino pyridine (5 mg, 0.037 mmol) in CH2Cl2 (20 mL), followed by reduction using NaBH4. As for the synthesis of [(piq)2Ir(L2)] encapsulated CdSe/ZnS QDs, namely Ir–CdSe/ZnS, the tri-n-butylphosphine (TBP)/tri-n-octylphosphine oxide (TOPO)-capped CdSe/ZnS QDs (10 mg) were dissolved in MeOH (15 mL) containing [(piq)2Ir(L2)] (50 mg, 0.05 mmol) at a pH value of ∼ 12, adjusted with tetramethylammonium hydroxide pentahydrate. The mixture was heated under reflux at 65 °C overnight, and then the reaction was terminated and the mixture allowed to cool to room temperature. Ir–CdSe/ZnS were then precipitated with diethyl ether. For further purification, the crude solid was washed with CH2Cl2 several times. Detailed synthetic procedures and characterization in each intermediate step are described in the supplementary information.
![(i) NaOEt; (ii) N2H4; (iii) HCl; (iv) [(piq)2IrCl]2; (v) 1. thiotic acid, DCC/DMAP, r.t., 72 h; 2. NaBH4, MeOH, r.t., 4 h; (vi) [(piq)2Ir(L2)], Me4N(OH), pH = 11, MeOH, reflux, 24 h.](/image/article/2006/CC/b517368j/b517368j-s1.gif) | ||
| Scheme 1 (i) NaOEt; (ii) N2H4; (iii) HCl; (iv) [(piq)2IrCl]2; (v) 1. thiotic acid, DCC/DMAP, r.t., 72 h; 2. NaBH4, MeOH, r.t., 4 h; (vi) [(piq)2Ir(L2)], Me4N(OH), pH = 11, MeOH, reflux, 24 h. | ||
Characterization of Ir–CdSe/ZnS was first performed with IR measurement. Figs. 1A and 1B depict the typical IR spectra of neat [(piq)2Ir(L2)] and Ir–CdSe/ZnS. In comparison, the resemblance in both spectral features and peak positions for most vibrational modes such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1450–1600 cm−1) and CN stretching (∼ 2000 cm−1) seems to guarantee a successful attachment of [(piq)2Ir(L2)] onto CdSe/ZnS QDs. The absence of S–H stretch band in the range of 2400–2600 cm−1 in Fig. 1B (see grey circle) firmly supports the formation of the sulfur–CdSe/ZnS bond. The 1H NMR of Ir–CdSe/ZnS is shown in the ESI. The associated 1H NMR peaks in Ir–CdSe/ZnS, except for the missing S–H peak at δ 1.5, are nearly identical to those of a pure [(piq)2Ir(L2)] sample (also see ESI), further supporting the attachment of [(piq)2Ir(L2)] on the surface of CdSe/ZnS.
C (1450–1600 cm−1) and CN stretching (∼ 2000 cm−1) seems to guarantee a successful attachment of [(piq)2Ir(L2)] onto CdSe/ZnS QDs. The absence of S–H stretch band in the range of 2400–2600 cm−1 in Fig. 1B (see grey circle) firmly supports the formation of the sulfur–CdSe/ZnS bond. The 1H NMR of Ir–CdSe/ZnS is shown in the ESI. The associated 1H NMR peaks in Ir–CdSe/ZnS, except for the missing S–H peak at δ 1.5, are nearly identical to those of a pure [(piq)2Ir(L2)] sample (also see ESI), further supporting the attachment of [(piq)2Ir(L2)] on the surface of CdSe/ZnS.
![FT-IR spectra of pure [(piq)2Ir(L2)] (A) and the [(piq)2Ir(L2)]-capped CdSe/ZnS QDs (B).](/image/article/2006/CC/b517368j/b517368j-f1.gif) | ||
| Fig. 1 FT-IR spectra of pure [(piq)2Ir(L2)] (A) and the [(piq)2Ir(L2)]-capped CdSe/ZnS QDs (B). | ||
Fig. 2 shows the absorption and emission spectra of [(piq)2Ir(L2)] and Ir–CdSe/ZnS. The spectral assignment of [(piq)2Ir(L2)] is straightforward, in which the lowest lying transition, at 570–600 nm with the absorption extinction of < 300 M−1cm−1, (see ESI) is assigned to the metal-to-ligand charge transfer (MLCT) in the triplet manifold. For comparison, the absorption of TOPO capped CdSe/ZnS QDs is also depicted; it exhibited an emission peak at ∼ 580 nm (not shown here) with a quantum efficiency of ∼ 0.42. Since Ir–CdSe/ZnS was prepared via a ligand exchange process from the TOPO capped CdSe/ZnS QDs, their similarity in size is expected. This viewpoint is supported by the TEM results, in which the average diameters of TOPO capped CdSe/ZnS and Ir–CdSe/ZnS, measured by TEM, were calculated to be 6.8 ± 0.7 and 7.0 ± 0.6 nm, respectively (see ESI). Since the diameter of the CdSe core was measured to be ∼ 3.8 nm, the thickness of ZnS was > 1.5 nm. As shown in Fig. 2, the absorption spectrum of Ir–CdSe/ZnS is apparently composed of the absorption profile of [(piq)2Ir(L2)] and CdSe/ZnSe QDs. The steady state emission of Ir–CdSe/ZnS in degassed MeOH consists of a distinct band maximized at 590 nm and a shoulder around 650 nm. Upon aeration, the 650 nm shoulder nearly disappeared, accompanied by a decrease of the overall emission intensity (see Fig. 2). However, the 590 nm peak position remained unchanged. In view of the spectral position and bandwidth, the 590 nm emission profile resembles that (580 nm) of the TOPO-capped CdSe/ZnS QDs. Thus, its assignment to the CdSe/ZnS emission seems unambiguous. The ∼ 10 nm red shift is possibly due to the different capping environment, i.e. TOPO and toluene versus [(piq)2Ir(L2)] and MeOH. Assuming that the spectrum obtained in the aerated solution is mainly attributed to the CdSe/ZnS emission, the spectrum acquired in the degassed solution can thus be well convoluted by a combination of the emission spectra of CdSe/ZnS (590 nm) and [(piq)2Ir(L2)] (610 nm). These results clearly indicate that the 610 nm emission, which is subject to drastic O2 quenching, is attributed to the phosphorescence of [(piq)2Ir(L2)]. Negligible interference (e.g. energy transfer) between CdSe/ZnS and [(piq)2Ir(L2)] chromophores is supported by the following experimental data. Upon monitoring at the emission wavelength of ∼ 750 nm, which solely originates from the [(piq)2Ir(L2)] emission, the excitation spectrum is identical with the absorption profile of the QDs-free [(piq)2Ir(L2)]. Furthermore, upon monitoring at e.g. 600 nm, the relaxation dynamics of Ir–CdSe/ZnS in degassed MeOH were composed of a fast component and a much slower decay component, the lifetimes of which were fitted to be 32 ns and 2.1 µs, respectively. Upon aeration, the 2.1 µs component was drastically reduced to ∼ 200 ns, while the fast component remained unchanged in either pre-exponential or decay time (i.e. ∼ 30 ns, see Fig. 3). One can thus safely conclude that the Förster type of resonance energy transfer is either very minor or even not operative in the Ir–CdSe/ZnS system.
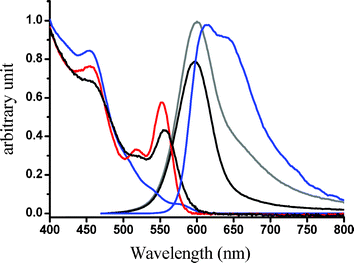 | ||
| Fig. 2 Absorption and emission spectra of Ir-complex (blue) and Ir–CdSe/ZnS QDs (grey for degassed, black for aerated) in MeOH; (red) absorption spectra of TOPO-capped CdSe/ZnS in toluene. | ||
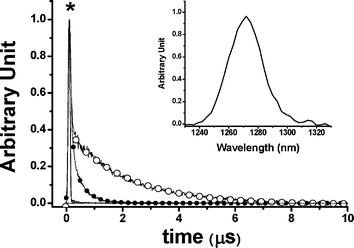 | ||
| Fig. 3 Decay dynamics at 650 nm for degassed (–○–) and aerated Ir–CdSe/ZnS (–●–) in MeOH. * denotes system response function. For aerated Ir-QDs: τ1 = 30 ns, τ2 = 200 ns; degassed Ir-QDs: τ1 = 32 ns, τ2 = 2.1 µs, λex = 450 nm. Inset: emission spectra of singlet oxygen upon exciting Ir–CdSe/ZnS in aerated MeOH (λex = 514 nm, Ar+ laser). | ||
The generation of 1O2 in the Ir–CdSe/ZnS system was supported by the observation of 1Δg (0) →
1Σ−g (0) 1273 nm emission upon exciting Ir–CdSe/ZnS in the aerated MeOH (see insert of Fig. 3). The assignment of 1O2 emission is unambiguous based on two observations. First, this 1273 nm emission disappeared upon degassing. Secondly, the lifetime of the emission revealed drastic solvent isotope dependence, being shifted from 25 µs in MeOH to ∼ 240 µs in CD3OD, consistent with a 1O2 electronic transition-solvent vibrational energy matching mechanism.12 We further made an attempt to estimate the yield of 1O21Δg (0) →
1Σ−g (0) 1273 nm emission. In this approach, the compound bis(triisobutylsiloxy) silicon-2,3-naphthalocyanine (SiINC) was used as a reference, of which the 1342 nm phosphorescence yield has been determined to be 7.47 × 10−5 in benzene.13 Under experimental conditions where the number of photons being absorbed by the Ir–CdSe/ZnS and SiINC are identical at e.g. 600 nm, the relative quantum yield of the 1O2 phosphorescence in MeOH with respect to that of the SiINC phosphorescence in THF was calculated on the basis of the following relationship
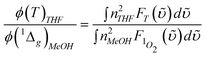
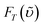 and
and 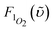 are the phosphorescence spectra of SiINC and O2 (1Δg), respectively. n denotes the refraction index of the solvent, which is 1.328 for MeOH and 1.401 for THF.14 As a result, the quantum yield of O2 (1Δg) 1273 nm emission was calculated to be 4.1 × 10−5 in MeOH. This value is ∼ 87% of the 1O2 sensitized by 1H-phenalen-1-one (PH) in the MeOH (∼ 4.7 × 10−5).15 Since the efficiency of PH sensitizing 1O2 in aerated solution is near unity,15b it is reasonable to conclude that the 1O2 production is ∼ 87% for Ir–CdSe/ZnS. In aerated MeOH, since the decay dynamics of Ir–CdSe/ZnS phosphorescence are dominated by the O2 quenching process (see Fig. 3), the O2 quenching efficiency is thus nearly equivalent to the ratio of [kaer (5 × 106 s−1) −
kdegas (4.76 × 105 s−1)] versuskaer and was estimated to be 90%. As a result, the efficiency of the 1O2 production is deduced to be as high as 97%.
are the phosphorescence spectra of SiINC and O2 (1Δg), respectively. n denotes the refraction index of the solvent, which is 1.328 for MeOH and 1.401 for THF.14 As a result, the quantum yield of O2 (1Δg) 1273 nm emission was calculated to be 4.1 × 10−5 in MeOH. This value is ∼ 87% of the 1O2 sensitized by 1H-phenalen-1-one (PH) in the MeOH (∼ 4.7 × 10−5).15 Since the efficiency of PH sensitizing 1O2 in aerated solution is near unity,15b it is reasonable to conclude that the 1O2 production is ∼ 87% for Ir–CdSe/ZnS. In aerated MeOH, since the decay dynamics of Ir–CdSe/ZnS phosphorescence are dominated by the O2 quenching process (see Fig. 3), the O2 quenching efficiency is thus nearly equivalent to the ratio of [kaer (5 × 106 s−1) −
kdegas (4.76 × 105 s−1)] versuskaer and was estimated to be 90%. As a result, the efficiency of the 1O2 production is deduced to be as high as 97%.
In conclusion, we have ingeniously designed an Ir–CdSe/ZnS system, in which the interplay between CdSe/ZnS QDs and [(piq)2Ir(L2)] chromophores is negligible. The system possesses a bifunctional property in that CdSe/ZnS QDs and [(piq)2Ir(L2)] act as an imaging center and a 1O2 sensitizing agent, respectively. For Ir–CdSe/ZnS in aerated MeOH, the quantum yield of the 590 nm CdSe/ZnS emission was determined to be 0.4, which is sufficiently high for application in imaging. As the next practical application, specific target agents can also be designed and co-anchored with [(piq)2Ir(L2)] ligand to CdSe/ZnS, among which a potential candidate should be folic acid because it binds to a receptor that several kinds of cancer cells produce in unusually large amounts.16 The resulting system is expected to be water soluble as well as to possess a three-in-one property, namely specific targeting, imaging, and 1O2 generation, which would greatly expand the usefulness of photodynamic therapy.
Notes and references
- (a) D. L. Klein, R. Roth, A. K. L. Lim, A. P. Alivisatos and P. L. McEuen, Nature, 1997, 389, 699 CrossRef CAS; (b) W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425 CrossRef CAS.
- (a) M. Gao, B. Richter, S. Kirstein and H. Mohwald, J. Phys. Chem. B, 1998, 102, 4096 CrossRef CAS; (b) J. Lee, V. C. Sundar, J. Heine, M. G. Bawendi and K. F. Jemsen, Adv. Mater., 2000, 12, 1102 CrossRef CAS; (c) S. Coe, W. Woo, M. Bawendi and V. Bulovic, Nature, 2002, 420, 800 CrossRef CAS; (d) N. Tessler, V. Medvedev, M. Kazes, S. Kan and U. Banin, Science, 2002, 295, 1506 CrossRef.
- (a) M. Bruchez, M. Moronne, P. Gin, W. Shimon and A. P. Alivisatos, Science, 1998, 281, 2013 CrossRef CAS; (b) W. C. W. Chan and S. Nie, Science, 1998, 281, 2016 CrossRef CAS; (c) D. Gerion, W. J. Parak, S. C. Williams, D. Zanchet, C. M. Micheel and A. P. Alivisatos, J. Am. Chem. Soc., 2002, 124, 7070 CrossRef CAS.
- A. K. Jeremiah, C. Netta and L. N. Jay, J. Phys. Chem. B, 2004, 108, 17042 CrossRef.
- (a) A. C. Samia, X. Chen and C. Burda, J. Am. Chem. Soc., 2003, 125, 15736 CrossRef CAS; (b) R. Bakalova, H. Ohba, Z. Zhelev, M. Ishikawa and Y. Baba, Nat. Biotechnol., 2004, 22, 1360 CrossRef CAS; (c) A. R. Clapp, I. L Medintz, J. M. Mauro, B. R. Fisher, M. G. Bawendi and H. Mattoussi, J. Am. Chem. Soc., 2004, 126, 301 CrossRef CAS; (d) I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435 CrossRef CAS; (e) B. R. Fisher, H.-J. Eisler, N. E. Stott and M. G. Bawendi, J. Phys. Chem. B, 2004, 108, 143 CrossRef CAS; (f) I. L. Medintz, S. A. Trammel, H. Mattoussi and J. M. Mauro, J. Am. Chem. Soc., 2004, 126, 30 CrossRef CAS; (g) D. M. Willard and A. Van Orden, Nat. Mater., 2003, 2, 575 CrossRef CAS.
- (a) B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759 CrossRef CAS; (b) E. G. Soltesz, S. Kim, R. G. Laurence, A. M. DeGrand, C. P. Parungo, D. M. Dor, L. H. Cohn, M. G. Bawendi, J. V. Frangioni and T. Mihaljevic, Ann. Thorac. Surg., 2005, 79, 269 CrossRef.
- (a) X. H. Gao and S. M. Nie, Trends Biotechnol., 2003, 21, 371 CrossRef CAS; (b) T. M. Jovin, Nat. Biotechnol., 2003, 21, 32 CrossRef CAS.
- (a) R. L. Morris, K. Azizuddin, M. Lam, J. Berlin, A. Nieminen, M. E. Kenney, A. C. S. Samia, C. Burda and N. L. Oleinick, Cancer Res., 2003, 63, 5194 CAS; (b) T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889 CrossRef CAS.
- (a) D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380 CrossRef CAS; (b) M. B. Vrouenraets, G. W. M. Visser, S. B. Snow and G. A. M. S. van Dongen, Anticancer Res., 2003, 23, 505 CAS.
- P. Reiss, J. Bleuse and A. Pron, Nano Lett., 2002, 2, 781 CrossRef CAS.
- F.-M. Hwang, H.-Y. Chen, P.-S. Chen, C.-S. Liu, Y. Chi, C.-F. Shu, F.-I. Wu, P.-T. Chou, S.-M. Peng and G.-H. Lee, Inorg. Chem., 2005, 44, 1344 CrossRef CAS.
- (a) J. R. Huster and G. B. Schuster, J. Am. Chem. Soc., 1983, 105, 5756 CrossRef CAS; (b) M. A. J. Rogers, J. Phys. Chem., 1983, 105, 6201.
- P. T. Chou, Y. C. Chen, S. J. Chen, M. Z. Lee, C. Y. Wei and T. C. Wen, J. Chin. Chem. Soc., 1998, 45, 503.
- T. J. Bruno and P. D. N. Svoronos, CRC Handbook of Basic Tables for Chemical Analysis, CRC Press, Boca Raton, FL, 1989, p. 89 Search PubMed.
- (a) K. I. Salokhiddinov, D. M. Dzhagarov, I. M. Byteva and G. P. Guinovich, Chem. Phys. Lett., 1980, 76, 85 CrossRef CAS; (b) R. Schmidt and E. Afshari, J. Phys. Chem., 1990, 94, 4377 CrossRef.
- (a) T Hasan, in: G. J. Dougherty, B. Henderson, editors, Photodynamic Therapy: Basic Principles and Clinical Applications, Marcel Dekker: New York, 1992, pp. 187–200 Search PubMed; (b) L. Strong, D. M. Yarmush and M. L. Yarmush, Ann. N.Y. Acad. Sci., 1994, 745, 297 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed syntheses and characterization and measurements. See DOI: 10.1039/b517368j |
| This journal is © The Royal Society of Chemistry 2006 |