Highly enantioselective DNA-based catalysis†
Gerard
Roelfes
*,
Arnold J.
Boersma
and
Ben L.
Feringa
*
Department of Organic Chemistry, Stratingh Institute, University of Groningen, Nijenborgh 4, Groningen, The Netherlands. Fax: +31 50 3634296; Tel: +31 50 3634235E-mail: J.G.Roelfes@rug.nl; B.L.Feringa@rug.nl
First published on 6th January 2006
Abstract
A new approach to DNA-based asymmetric catalysis is presented, which gives rise to very high enantioselectivities (up to 99% ee) in the copper catalyzed Diels–Alder reaction in water.
DNA, with its characteristic right-handed helix structure, has become one of the icons of modern science. Due to its versatile architecture, DNA represents an ideal scaffold for the construction of catalytic systems, known as deoxyribozymes. These DNAzymes have found many applications, primarily due to their ability to hydrolyze RNA.1 Because of its inherent chirality, DNA is an attractive scaffold for the design of enantioselective catalysts. However, compared to RNAzymes,2 DNAzymes posses, as yet, a more limited catalytic repertoire, as is illustrated by the fact that, to date, DNAzymes capable of catalyzing C–C bond forming reactions have not been reported.
Recently, we introduced a novel DNA-based asymmetric catalysis concept based on the modular assembly of a DNA-based catalyst, i.e. DNA with extended catalytic functionality, attached in a non-covalent manner.3 This catalytic ensemble comprises a copper complex of a non-chiral ligand, which incorporates a metal binding site, a spacer and a covalently attached intercalator, i.e. 9-amino acridine. As a result, the active Cu(II) centre is brought into proximity of the chiral environment of the DNA double helix, allowing for a transfer of chirality from DNA to the reaction product. With this approach enantiomeric excess (ee) for copper catalyzed Diels–Alder reactions of ∼50% for the major (endo) isomer and up to 90% for the minor (exo) isomer could be achieved. Although the ligand in the acridine-based systems is achiral, the corresponding copper complex is chiral, which raises the possibility that the transfer of chirality from DNA to the catalyzed reaction in this system proceeds via the preferred formation of one of the enantiomers of the copper complex. This concept of converting of an achiral ligand–metal complex to a chiral complex by an exogenous chiral source has been well established in asymmetric catalysis.4
In the present communication, we introduce a new and simplified approach to DNA-based catalysts (Scheme 1), in which chirality is transferred directly from DNA to the catalyzed reactions. For this purpose achiral copper complexes based on bidentate ligands known to bind to DNA (Fig. 1) are employed. Since in this approach the metal binding site and the DNA ‘anchor’ are integrated into one moiety, a spacer is no longer required. As a result the reactive Cu(II) centre is brought into even closer contact with the double helix. This new approach gives rise to a dramatic increase in enantioselectivity of the Cu(II) catalyzed asymmetric Diels–Alder reaction compared to our earlier system.
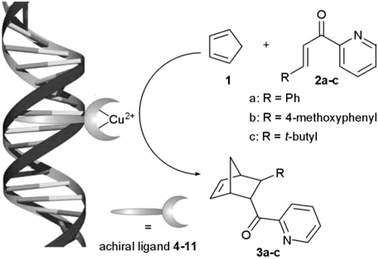 | ||
| Scheme 1 Schematic representation of the asymmetric Diels–Alder reaction catalyzed by a DNA-based catalyst. | ||
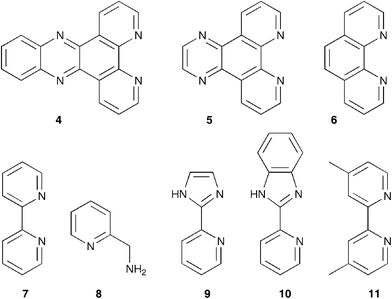 | ||
| Fig. 1 Ligands used in the present study. | ||
The DNA-based catalysts investigated initially, in this study, consist of salmon testes DNA (st-DNA) bound to a series of Cu(II) complexes based on simple achiral polyaromatic bidentate ligands, e.g. dipyrido[3,2-a∶2′,3′-c]phenazine (dppz, 4), dipyrido[2,2-d∶2′,3′-f]quinoxaline (dpq, 5), phenanthroline (phen, 6) and 2,2′-bipyridine (bipy, 7) (Fig. 1).5 The corresponding Cu(II) complexes, [Cu(L)(NO3)2], where L = 4–7, exhibit moderate to strong binding to DNA, with binding constants ranging from 9.3 × 103 M−1 to 8 × 105 M−1 (Table 1, entries 1–4). That the complexes are embedded in the chiral environment of the DNA is evident from the induced CD effect observed.†
| Entry | L | K b (DNA)/M−1 | Dienophile | Endo ∶ exo | ee (%)b |
|---|---|---|---|---|---|
| a All experiments were carried out with st-DNA (1.3 mg mL−1), 0.3 mM [Cu(L)(NO3)2], 1 mM 2a–c and 15 mM cyclopentadiene in MOPS buffer (20 mM pH 6.5) for 3 days at 5 °C, unless noted otherwise. b For the endo isomer. c 0.05 mM [Cu(L)(NO3)2]. d Calf thymus DNA. e Conversion ∼50%. f Cu(II) complex prepared in situ. g 2 eq. with respect to Cu(II). h At room temperature. | |||||
| 1 | 4 | 8 ± 3 × 105 | 2a | 96 ∶ 4 | 49 |
| 2 | 5 | 7.2 ± 1.2 × 104 | 2a | 95 ∶ 5 | 61 |
| 3 | 6 | 1.3 ± 0.1 × 104 | 2a | 96 ∶ 4 | 73 |
| 4 | 7 | 9.4 ± 0.3 × 103 | 2a | 98 ∶ 2 | 90 |
| 5c | 7 | — | 2a | 97 ∶ 3 | 89 |
| 6d | 7 | — | 2a | 97 ∶ 3 | 89 |
| 7 | 7 | — | 2b | 97 ∶ 3 | 92 |
| 8e | 7 | — | 2c | 94 ∶ 6 | 83 |
| 9f | 8 | — | 2a | 93 ∶ 7 | <5 |
| 10f | Pyg | — | 2a | 92 ∶ 8 | 6 |
| 11h | — | — | 2a | 95 ∶ 5 | −10 |
| 12 | 9 | 5.2 ± 0.3 × 103 | 2a | >99 ∶ 1 | 91 |
| 13 | 10 | 1.5 ± 0.1 × 104 | 2a | 98 ∶ 2 | 92 |
| 14e | 10 | — | 2b | >99 ∶ 1 | 95 |
| 15 | 10 | — | 2c | 98 ∶ 2 | 90 |
| 16 | 11 | 1.12 ± 0.02 × 104 | 2a | >99 ∶ 1 | 99 |
| 17c | 11 | — | 2a | 99 ∶ 1 | 98 |
| 18 | 11 | — | 2b | >99 ∶ 1 | >99 |
| 19 | 11 | — | 2c | >99 ∶ 1 | 97 |
The Diels–Alder reaction of cyclopentadiene (1) with aza-chalcone 2a catalyzed by DNA-based catalysts, assembled from st-DNA‡ and [Cu(L4–7)(NO3)2], was performed at 5 °C for 3 days. This ensured >80% conversion in all reactions, with the Diels–Alder product 3a being the sole product formed, according to 1H-NMR.§ The endo isomer of 3a, which was produced almost exclusively (≥95%), was obtained with ee’s ranging from 49% for [Cu(dppz)(NO3)2] to a remarkable 90% for [Cu(bipy)(NO3)2] (entries 1–4). This is considerably higher than those obtained previously with either the acridine-based systems or Cu(II)–amino acid complexes.6 Since, due to the symmetric, planar nature of ligands 4–6, the Cu(II) complexes are achiral, these results demonstrate the direct transfer of chirality from DNA to the catalyzed Diels–Alder reaction.¶
Ligands 4–7 provide a similar coordination environment to the Cu(II) ion and, hence, the relationship between the DNA binding strength of the complexes and the enantioselectivity observed in DNA-based catalysis can be established readily by comparison of the complexes' catalytic properties. Interestingly, within this homologous series, an inverse correlation was observed between the DNA binding strength of the Cu(II) complex and the ee in the product; the weakest DNA-binder in this series, [Cu(bipy)(NO3)2], provided the highest ee (Table 1). Catalysts based on 2-aminomethylpyridine (8) or pyridine (py) itself did not provide significant enantioselectivity in the reaction (entries 9, 10), whereas in the absence of ligand (i.e. only Cu(NO3)2 and DNA), 10% ee of the opposite enantiomer was obtained (entry 11). These results demonstrate that a copper complex capable of binding to DNA, although not necessarily with high affinity, is an absolute requirement in obtaining high enantioselectivity.
In view of the excellent results obtained with 2,2′-bipyridine, a series of bidentate ‘bipyridine-type’ ligands (9–11) were investigated. The pyridyl–imidazole ligands 9 and 10 provided up to 92% ee for 3a and 95% in case of 3b (entries 12–14). However, the best results were obtained with the Cu2+ complex of 4,4′-dimethyl-2,2′-bipyridine (dmbipy, 11), i.e. complete endo selectivity and virtually complete enantioselectivity (entry 16). Upon lowering the concentration of [Cu(dmbipy)](NO3)2 the reaction became somewhat slower, i.e. ∼45% vs. 28% conversion at 30 and 5 mol% catalyst, respectively, after 5.5 h. However, in both cases the reaction is essentially complete within 24 h and the enantioselectivity was not affected noticeably (entries 16, 17).
When the ratio of DNA base pairs : [Cu(bipy)(NO3)2] was lowered, either by reducing the DNA concentration or increasing the complex concentration, a slight decrease in ee was observed.† Considering the DNA-binding strength of [Cu(bipy)(NO3)2] and the number of available binding sites, this is not unexpected: at higher complex ∶ DNA ratios relatively more of the copper complex will remain unbound and be available, in solution, to catalyze the Diels–Alder reaction in a racemic fashion, resulting in a lower overall ee. This underscores further the need for the reaction to take place in close proximity to the DNA in order to obtain enantioselectivity. Performing the reaction at a larger scale gave rise to 67% conversion after 2 nights, resulting in 50% isolated yield of the pure Diels–Alder product 3a after column chromatography.||
The R group on the dienophile was found to be amenable to variation. Substrates 2b and 2c were converted efficiently to the corresponding Diels–Alder product with very high enantioselectivity (entries 7, 8, 14, 15, 18 and 19), albeit with lower conversion for 2b and with considerable amounts of a side-product, in the case of 2c. This side-product was the result of the Michael addition of water to 2c and was formed as a racemate. To date the keto-pyridyl group of the dienophile has proven to be essential to obtain enantioselectivity. Bidentate binding of the dienophile appears to be a general requirement of Cu(II) catalyzed Diels–Alder reactions, especially so in water.7 However, other bidentate binding dienophiles, like the Evans-type N-acryloyl oxazolidinones, do not compete effectively with H2O for the free coordination sites on copper, resulting in no observable catalytic effect and, hence, no enantioselectivity.8
Since the chirality is derived directly from the DNA (vide supra), it is clear that the enantioselectivity in the reaction will show a strong dependence on the precise nature of the interaction between DNA and the Cu(II) complex. [Cu(dppz)(NO3)2] and [Cu(dpq)(NO3)2] bind to DNA through intercalation.4 However, for the other complexes the precise DNA binding modes, i.e. intercalation, groove binding or a combination of both, are at present unknown. Nevertheless, some important observations can be made. It is apparent that the highest ee’s were achieved with complexes that exhibit moderate DNA-binding strength, i.e.Kb ∼ 103–104 M−1. This suggests some flexibility in the binding geometry of the complex is beneficial, possibly to accommodate the active, dienophile bound complex in a more favorable orientation in the DNA groove. Moreover, the optimum results were obtained with the biaryl type ligands, 7 and 9–11 that form Cu(II) complexes in which the ligand is known to be slightly twisted around the central C2–C2′ bond.9 A slight twist could lead to a better fit in the DNA helix, which would lead to less unwinding of the DNA helix, resulting in a more compact DNA-groove.10 Thus the probability of the diene approaching selectively from one pro-chiral face of the Cu(II) bound dienophile is increased, leading to higher enantioselectivity. Ultimately, to elucidate the mechanism by which chiral information is transferred from DNA to the product a detailed understanding of the mode of binding of the Cu(II)–dienophile complex is essential. Currently, an extensive spectroscopic study is underway to gain more insight into the structural factors involved.
In conclusion, this new and simple approach to DNA-based asymmetric catalysis enables very high enantioselectivities in the Cu(II) catalyzed Diels–Alder reaction to be achieved. Through this approach the assembly of DNA-based catalysts in a modular fashion from DNA and achiral copper complexes of simple and readily available achiral pyridine-based ligands is realized. The results presented demonstrate that the DNA is the source of chirality in these reactions and that the close contact between DNA and the copper complex allows for direct transfer of this chirality to the catalyzed reaction. The virtually complete regioselectivity (up to >99% endo) and excellent enantioselectivity (up to 99% ee) of the catalyzed Diels–Alder reactions in water, reported here, are testament to the potential of DNA-based asymmetric catalysis.
The authors thank the Netherlands Organization for Scientific Research (NWO) and the NRSC-Catalysis for financial support and Dr W. Browne for valuable discussions.
Notes and references
- S. K. Silverman, Org. Biomol. Chem., 2004, 2, 2701–2706 RSC; A. Peracchi, ChemBioChem, 2005, 6, 1316–1322 CrossRef CAS; Y. Lu, Chem.–Eur. J., 2002, 8, 4589–4596 CrossRef CAS; J. P. May, R. Ting, L. Lermer, J. M. Thomas, Y. Roupioz and D. M. Perrin, J. Am. Chem. Soc., 2004, 126, 4145–4156 CrossRef CAS.
- S. Fusz, A. Eisenführ, S. G. Srivatsan, A. Heckel and M. Famulok, Chem. Biol., 2005, 12, 941–950 CrossRef CAS; G. F. Joyce, Annu. Rev. Biochem., 2004, 73, 791–836 CrossRef CAS; F. Stuhlmann and A. Jäschke, J. Am. Chem. Soc., 2002, 124, 3238–3244 CrossRef CAS; B. Seelig, S. Keiper, F. Stuhlmann and A. Jäschke, Angew. Chem., Int. Ed., 2000, 39, 4576–4579 CrossRef CAS; T. M. Tarasow, S. L. Tarasow and B. E. Eaton, J. Am. Chem. Soc., 2000, 122, 1015–1021 CrossRef CAS; T. M. Tarasow, S. L. Tarasow and B. E. Eaton, Nature, 1997, 389, 54–57 CrossRef CAS.
- G. Roelfes and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 3230–3232 CrossRef CAS.
- P. J. Walsh, A. E. Lurain and J. Balsells, Chem. Rev., 2003, 103, 3297–334 CrossRef CAS.
- A. K. Patra, S. Dhar, M. Nethaji and A. R. Chakravarty, Dalton Trans., 2005, 896–902 RSC; M. Navarro, E. J. Cisneros-Fajardo, A. Sierralta, M. Fernández-Mestre, P. Silva, D. Arrieche and E. Marchán, JBIC, J. Biol. Inorg. Chem., 2003, 8, 401–408 CAS; K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777–2795 CrossRef CAS.
- S. Otto and J. B. F. N. Engberts, J. Am. Chem. Soc., 1999, 121, 6798–6806 CrossRef CAS; S. Otto, G. Boccaletti and J. B. F. N. Engberts, J. Am. Chem. Soc., 1998, 120, 4238–4239 CrossRef CAS.
- T. Rovis and D. A. Evans, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, New York, 2001, vol. 50, ch. 1, pp. 89–129 Search PubMed; S. Otto, F. Bertoncin and J. B. F. N. Engberts, J. Am. Chem. Soc., 1996, 118, 7702–7707 Search PubMed.
- K. Manabe, Y. Mori and S. Kobayashi, Tetrahedron, 1999, 55, 11203–11208 CrossRef CAS.
- G. A. Barclay, C. H. L. Kennard and B. F. Hoskins, J. Chem. Soc., 1963, 5691–5699 RSC; I. M. Procter and F. S. Stephens, J. Chem. Soc. A, 1969, 1248–1255 RSC; L. P. Battaglia, M. Belicchiferrari, A. Bonamartinicorradi, G. Gasparrifava, C. Pelizzi and M. E. Vidonitani, J. Chem. Soc., Dalton Trans., 1976, 2197–2202 RSC.
- L. Strekowski, J. L. Mokrosz, F. A. Tanious, R. A. Watson, D. Harden, M. Mokrosz, W. D. Edwards and W. D. Wilson, J. Med. Chem., 1988, 31, 1231–1240 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for catalytic experiments, synthesis and characterization of ligands and complexes, Kb determinations, CD spectra and tables with results of catalytic experiments. See DOI: 10.1039/b516552k |
| ‡ Calf thymus DNA proved equally effective in this reaction compared with st-DNA (Table 1, entry 6), demonstrating that this system is not dependent on the DNA source. |
| § In the absence of DNA generally a slightly lower conversion was obtained, whereas hardly any reaction was observed with only DNA and no copper complex (see supplementary information†). |
| ¶ Even though [Cu(bipy)(NO3)2] is chiral as a result of the small twist between the two pyridyl groups, this twist is generally quite small and does not lead to significantly different geometries around the Cu(II) centre. Therefore, it is unlikely that the chirality in the complex is responsible for the observed enantioselectivity and, hence, in this case most likely also is direct chirality transfer from DNA. |
| || Reaction performed on a 50 mL scale, with 0.3 mM [Cu(bipy)(NO3)2], 1.8 mM dienophile 2a and 45 mM cyclopentadiene, for 2 days at 5 °C. |
| This journal is © The Royal Society of Chemistry 2006 |