Double functionalisation of carbon nanotubes for multimodal drug delivery†
Giorgia
Pastorin
a,
Wei
Wu
a,
Sébastien
Wieckowski
a,
Jean-Paul
Briand
a,
Kostas
Kostarelos
b,
Maurizio
Prato
*c and
Alberto
Bianco
*a
aInstitut de Biologie Moléculaire et Cellulaire, UPR9021 CNRS, Immunologie et Chimie Thérapeutiques, 67084, Strasbourg, France. E-mail: A.Bianco@ibmc.u-strasbg.fr; Fax: +33 388 610680; Tel: +33 388 417088
bCentre for Drug Delivery Research, The School of Pharmacy, University of London, 29-39 Brunswick Square, London, UK WC1N 1AX
cDipartimento di Scienze Farmaceutiche, Università di Trieste, 34127, Trieste, Italy. E-mail: prato@units.it
First published on 13th February 2006
Abstract
Multi-walled carbon nanotubes have been covalently functionalised via 1,3-dipolar cycloaddition of azomethine ylides with orthogonally protected amino functions that can be selectively deprotected and subsequently modified with drugs and fluorescent probes.
Functionalisation of carbon nanotubes (CNT) is a key step for the integration of this new material into different systems for technological and biomedical applications.1–8 One of the most powerful approaches for rendering carbon nanotubes soluble in a wide range of solvents is the covalent functionalisation of their side-walls and tips.9–11 As a consequence, functionalised carbon nanotubes (f-CNT) are emerging as novel components in nanovector formulations for the delivery of therapeutic molecules.5–8f-CNT loaded with different peptides, proteins and nucleic acids are able to deliver their cargos into cells.12–20 Covalent functionalisation of CNT with drug (e.g. anticancer, antiviral or antibacterial agents) molecules is instead a field of research still poorly explored.21 The development of functionalised carbon nanotubes to target and be uptaken by specific cell populations without collateral consequences for healthy tissues would be of fundamental importance for example in cancer treatment.22,23 The molecular targeting of carbon nanotube delivery systems derivatised with a therapeutic agent is possible if an active recognition moiety is simultaneously present at the surface of the nanocarrier.23 In addition, attachment of a fluorescent molecule would provide optical signals for imaging and localisation of the CNT–drug conjugates. Therefore, multiple functionalisation of CNT is of particular interest for multimodal delivery of anticancer agents.
In this study we describe a straightforward methodology for the introduction of two orthogonally protected amino groups on the sidewalls of CNT, subsequently derivatised with fluorescein isothiocyanate (FITC) and methotrexate (MTX). MTX is a drug widely used against cancer, however, it suffers from low cellular uptake.24,25 Its conjugation to CNT represents a promising approach to overcome its limited cellular uptake by enhancing its internalisation via the f-CNT.15 For this purpose we exploited the 1,3-dipolar cycloaddition of azomethine ylides recently reported by our group.26,27 Diaminotriethylene glycol was initially mono-protected as tert-butyloxycarbonyl (Boc) as previously reported,27 or as benzyloxycarbonyl (Z) (see ESI†). Both derivatives were subsequently reacted with benzyl- and tert-butylbromoacetate, respectively. Removal of the protection at the carboxylic functions using catalytic hydrogenolysis or acid treatment afforded the N-substituted glycines that were used for the 1,3-dipolar cycloaddition to multi-walled carbon nanotubes (MWNT) in the presence of paraformaldehyde at 130 °C in DMF (Scheme 1).
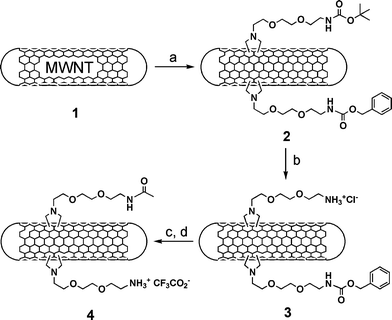 | ||
| Scheme 1 (a) R–NHCH2COOH/(CH2O)n in DMF, 130 °C; (b) HCl 4 M in dioxane; (c) Ac2O; (d) TFA/TMSOTf/para-cresol. R = Boc–NH(CH2CH2O)2–CH2CH2– and Z–NH(CH2CH2O)2–CH2CH2–. | ||
The reaction was conducted in the concomitant presence of an equimolar amount of both amino acids. After three days unreacted CNT were separated by centrifugation and f-MWNT were isolated by extraction with DCM (dichloromethane). Doubly f-MWNT were characterised by TEM and 1H NMR. In particular, the NMR analysis allowed observation of the resonances of the Z phenyl and methylene protons at 7.30 and 5.06 ppm, respectively, and Boc group at 1.39 ppm (see ESI†). Integration of these signals indicated the presence of an almost equal amount of the two moieties on the sidewalls of f-CNT. TEM allowed observation of tubes and helped determine their diameter and length characteristics (see ESI†). These two quasi-orthogonal protecting groups were removed selectively using different conditions. Boc was cleaved first by treating MWNT 2 with HCl in dioxane. After liberation of the amino functions, we measured an amount of 0.35 mmol g−1 of NH2 (Table 1).
| Cleavage conditions | HCl/dioxane | TFA/TMSOTf/para-cresol | Hydrazine/EtOH |
|---|---|---|---|
| a Determined by the quantitative Kaiser test. b The number in parentheses corresponds to the f-MWNT reported in Scheme 1. c Amount of free amines for f-MWNT derived from 5 (Scheme 2) after Boc or Pht cleavage, respectively. | |||
| Loading (mmol g−1) a | 0.35 (3)b | 0.35 (4)b | — |
| Loading (mmol g−1) a | 0.30c | — | 0.65c |
Before unmasking of the second protecting group, we blocked the free amines by direct acetylation (Scheme 1). Z group was then cleaved using a mixture of trifluoroacetic acid, trimethylsilyl trifluoromethanesulfonate and para-cresol (TFA/TMSOTf/para-cresol). Following precipitation and washings, we measured a loading of 0.35 mmol g−1 of free amines (Table 1). This type of cleavage sequence is required by the Boc/Z protecting strategy, which implies two strong acid treatments for the removal of Boc and Z, respectively. This approach is somehow of limited application, since for further modifications following deprotection of Boc not all molecules are stable to the strong conditions used for Z cleavage. For this reason, a second strategy based on the complete orthogonality for the protection of the amino functions was explored. In this case, we prepared a mono-phthalimide (Pht) protected diaminotriethylene glycol, which was then reacted with benzylbromoacetate (see ESI†). The carboxylic function was liberated and the N-glycine bearing the Pht was mixed with an equivalent amount of the Boc-protected analogue and added simultaneously to the MWNT (Scheme 2).
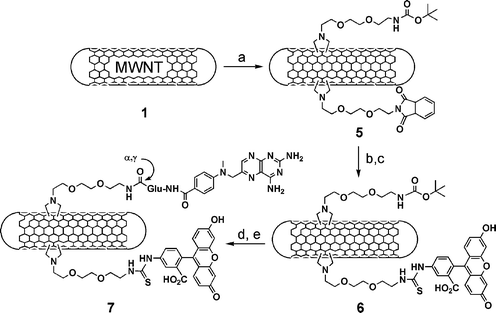 | ||
| Scheme 2 (a) R–NHCH2COOH/(CH2O)n in DMF, 130 °C; (b) hydrazine in EtOH; (c) FITC in DMF; (d) HCl 4 M in dioxane; (e) MTX, Bop/DIEA in DMF. R = Boc–NH(CH2CH2O)2–CH2CH2– and Pht–N(CH2CH2O)2–CH2CH2–. | ||
MWNT 5 were observed by TEM (Fig. 1) and characterised again by 1H NMR. Boc and Pht proton chemical shifts are present at around 1.40 and 7.00 ppm, respectively (see ESI†). The cleavage of Boc was performed using the acid conditions employed for MWNT 2, while Pht was removed using a solution of hydrazine in ethanol at room temperature under argon. The quantity of the free amino groups is reported in Table 1.
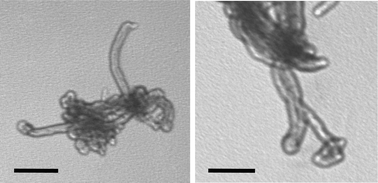 | ||
| Fig. 1 TEM images of MWNT 5 and 7. The scale bar corresponds to 200 nm. | ||
To prove the feasibility of double functionalisation of carbon nanotubes, we first removed Pht and labelled the tubes with fluorescein isothiocyanate (Scheme 2). Then, MWNT 6 were treated with HCl to eliminate the Boc group and coupled MTX.28 MTX was activated at the carboxylic groups, using BOP [benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate], and successfully coupled via the α or γ COOH to the free amino groups of Boc-deprotected MWNT 6. The negative Kaiser text confirmed the completeness of the reaction. The conjugate 7 was characterised using TEM (Fig. 1) and UV-Vis spectroscopy (see ESI†). TEM analysis of the conjugate 7 showed morphology very similar to MWNT 5.
We have subsequently evaluated the capacity of the bifunctional MWNT 7 to penetrate into the cells by epifluorescence and confocal microscopy. Human Jurkat T lymphocytes were cultured using RPMI 1640 medium at 37 °C. MWNT 7 were added to the cells at room temperature in a range of concentration between 0.05 and 5 µg ml−1, and the cells were incubated at 37 °C for 1 h. After this period, the cells were carefully rinsed, mounted on a microscope slide and observed under the microscope. Indeed, the presence of the fluorescence probe on the tubes has allowed the analysis of its internalisation and intracellular distribution. Fig. 2A–C shows the epifluorescence and confocal microscopy images of the Jurkat cells after being treated with 5 µg ml−1 of MWNT 7. It is evident that MWNT 7 accumulates into the cytoplasm (Fig. 2B). The confocal analysis (Fig. 2C) clearly shows the presence of the labelled compound inside the cell localised around the nuclear membrane. We finally studied the dose and time dependence of the internalisation process. Fig. 2D shows the flow cytometry analysis of the cells treated with three different amounts of MWNT 7 for 24 hours. It is clear that the observed fluorescence signal is proportional to the dose. The rapid internalisation of the MTX by carbon nanotubes will be particularly advantageous for an improved efficiency of the drug action. In addition, we have measured the effect of time on the cell internalisation using different doses of MWNT 7 (see ESI, Fig. S9†). The process was followed between 0 and 24 hours. Again, a fluorescence shift is strictly correlated to the dose with a fast kinetics of uptake for the highest concentration of 5 µg ml−1. The cell internalisation is essentially exerted by the CNT independently from the type of functional groups covalently linked to their external surface since MWNT 6 without the Boc group, precursor of MWNT 7, showed the same behaviour as 7 (see ESI, Fig. S7–S9†).
![Bright field (A), epifluorescence (B) and confocal (C) images of Jurkat cells incubated for 1 h at 37 °C with 5 µg ml−1 of MWNT 7. (D) Dose-response of the internalisation after incubation of Jurkat cells for 24 h at 37 °C with increasing amount of MWNT 7 [0.05 (thick grey line), 0.5 (thin black line) and 5 (thick black line) µg ml−1]. FL1-H corresponds to FITC intensity.](/image/article/2006/CC/b516309a/b516309a-f2.gif) | ||
| Fig. 2 Bright field (A), epifluorescence (B) and confocal (C) images of Jurkat cells incubated for 1 h at 37 °C with 5 µg ml−1 of MWNT 7. (D) Dose-response of the internalisation after incubation of Jurkat cells for 24 h at 37 °C with increasing amount of MWNT 7 [0.05 (thick grey line), 0.5 (thin black line) and 5 (thick black line) µg ml−1]. FL1-H corresponds to FITC intensity. | ||
In conclusion, we have developed a novel strategy for the functionalisation of carbon nanotubes with two different molecules using the 1,3-dipolar cycloaddition of azomethine ylides. We have explored two alternative routes, which allowed introduction of a fluorescent probe and an anticancer agent around the CNT sidewalls. Controlled multifunctionalisation of CNT will certainly open new perspectives in the field of medical applications of f-CNT. For example, the attachment of molecules that will target specific receptors on tumour cells will help improve the response to anticancer agents. We are currently studying the cytotoxic activity of MTX conjugated to the fluorescent carbon nanotubes.
This work has been supported by CNRS, MRNT (Programme GenHomme 2003), University of Trieste and MIUR (PRIN 2004, prot. 2004035502). G. P. and W. W. are recipients of a fellowship from MNRT. The authors wish to thank Sylvie Fournel for fruitful discussion and comments on the paper. Confocal images were collected at the Microscopy Facility Plate-form of the Institut de Biologie Moléculaire des Plantes (IBMP, Strasbourg, France).
Notes and references
- Special issue on carbon nanotubes, Acc. Chem. Res., 2002, 35, 997 CrossRef.
- Y. Lin, S. Taylor, H. Li, K. A. Fernando, L. Qu, W. Wang, L. Gu, B. Zhou and Y.-P. Sun, J. Mater. Chem., 2004, 14, 527 RSC.
- C. R. Martin and P. Kohli, Nat. Rev. Drug Discovery, 2003, 2, 29 CrossRef CAS.
- J. Bradbury, Lancet, 2003, 362, 1984 CrossRef.
- A. Bianco, K. Kostarelos, C. D. Partidos and M. Prato, Chem. Commun., 2005, 571 RSC.
- K. Kostarelos, L. Lacerda, C. D. Partidos, M. Prato and A. Bianco, J. Drug Delivery Sci. Technol., 2005, 15, 41 Search PubMed.
- A. Bianco, Expert Opin. Drug Delivery, 2004, 1, 57 Search PubMed.
- A. Bianco and M. Prato, Adv. Mater., 2003, 15, 1765 CrossRef.
- D. Tasis, N. Tagmatarchis, V. Georgakilas and M. Prato, Chem.–Eur. J., 2003, 9, 4000 CrossRef CAS.
- A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853 CrossRef CAS.
- C. A. Dyke and J. M. Tour, Chem.–Eur. J., 2004, 10, 812 CrossRef CAS.
- N. W. Shi Kam, T. C. Jessop, P. A. Wender and H. Dai, J. Am. Chem. Soc., 2004, 126, 6850 CrossRef CAS.
- D. Pantarotto, J.-P. Briand, M. Prato and A. Bianco, Chem. Commun., 2004, 16 RSC.
- N. W. Shi Kam and H. Dai, J. Am. Chem. Soc., 2005, 127, 6021 CrossRef CAS.
- D. Pantarotto, C. D. Partidos, R. Graff, J. Hoebeke, J.-P. Briand, M. Prato and A. Bianco, J. Am. Chem. Soc., 2003, 125, 6160 CrossRef CAS.
- D. Pantarotto, C. D. Partidos, J. Hoebeke, F. Brown, E. Kramer, J.-P. Briand, S. Muller, M. Prato and A. Bianco, Chem. Biol., 2003, 10, 961 CrossRef CAS.
- D. Pantarotto, R. Singh, D. McCarthy, M. Erhardt, J.-P. Briand, M. Prato, K. Kostarelos and A. Bianco, Angew. Chem., Int. Ed., 2004, 43, 5242 CrossRef CAS.
- R. Singh, D. Pantarotto, D. McCarthy, O. Chaloin, J. Hoebeke, C. D. Partidos, J.-P. Briand, M. Prato, A. Bianco and K. Kostarelos, J. Am. Chem. Soc., 2005, 127, 4388 CrossRef CAS.
- A. Bianco, J. Hoebeke, S. Godefroy, O. Chaloin, D. Pantarotto, J.-P. Briand, S. Muller, M. Prato and C. D. Partidos, J. Am. Chem. Soc., 2005, 127, 58 CrossRef CAS.
- N. W. S. Kam, K. Liu and H. Dai, J. Am. Chem. Soc., 2005, 127, 12492 CrossRef CAS.
- W. Wu, S. Wieckowski, G. Pastorin, M. Benincasa, C. Klumpp, J.-P. Briand, R. Gennaro, M. Prato and A. Bianco, Angew. Chem., Int. Ed., 2005, 44, 6358 CrossRef CAS.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161 CrossRef CAS.
- N. W. Shi Kam, M. O'Connel, J. A. Wisdom and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11600 CrossRef CAS.
- R. Pignatello, S. Guccione, S. Forte, C. Di Giacomo, V. Sorrenti, L. Vicari, G. Uccello Barretta, F. Balzano and G. Puglisi, Bioorg. Med. Chem., 2004, 12, 2951 CrossRef CAS.
- A. Quintana, E. Raczka, L. Piehler, I. Lee, A. Myc, I. Majoros, A. K. Patri, T. Thomas, J. Mule and J. R. Baker, Jr., Pharm. Res., 2002, 19, 1310 CrossRef CAS.
- V. Georgakilas, K. Kordatos, M. Prato, D. M. Guldi, M. Holzinger and A. Hirsch, J. Am. Chem. Soc., 2002, 124, 760 CrossRef CAS.
- V. Georgakilas, N. Tagmatarchis, D. Pantarotto, A. Bianco, J.-P. Briand and M. Prato, Chem. Commun., 2002, 3050 RSC.
- A. Nagy, B. Szoke and A. V. Schally, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6373 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details. See DOI: 10.1039/b516309a |
| This journal is © The Royal Society of Chemistry 2006 |