Rapid formation of amides via carbonylative coupling reactions using a microfluidic device
Philip W.
Miller
a,
Nicholas J.
Long
a,
Andrew J.
de Mello
a,
Ramon
Vilar
b,
Jan
Passchier
c and
Antony
Gee
c
aDepartment of Chemistry, Imperial College London, London, UK SW7 2AZ. E-mail: n.long@imperial.ac.uk; Fax: +44 (0)20 75945804; Tel: +44 (0)20 75945781
bInstitute of Chemical Research of Catalonia (ICIQ), Avgda. Paisos Catalans 16, 43007 Tarragona, Spain. E-mail: rvilar@iciq.es; Fax: +34 977 920 228; Tel: +34 977 920 212
cPET Imaging Division, Translational Medicine and Genetics, GlaxoSmithKline, Academic Centre for Clinical Investigation, Addenbrooke's Hospital, Hills Road, Cambridge, UK CB2 2GG
First published on 15th December 2005
Abstract
Carbonylative cross-coupling reactions of arylhalides to form secondary amides were rapidly carried out on a glass-fabricated microchip—the first time a microstructured device has been used to perform a gas–liquid carbonylation reaction.
Microstructured devices are finding ever more innovative applications in chemical synthesis and chemical engineering.1 A key feature of such devices is their increased surface area-to-volume ratio which is a direct result of their decrease in physical size. Specific surfaces of microstructured devices can be as high as 50
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 m−3 whereas conventional laboratory apparatus does not usually exceed 1000 m2 m−3. A consequence of this increase in specific surface is the enhancement of mass and heat transport in the system. Microreactors specifically designed for multiphase gas–liquid and gas–liquid–solid reactions have been used to carry out a range of reactions, including oxidation,2 fluorination,3 nitration4 and hydrogenation5 reactions, all of which have shown enhanced rates, yields and/or selectivities, compared to their corresponding batch reactions. These improvements are attributed to the increased interfacial contact area generated between the phases within the reactor. The advantages of using microreactors for gas–liquid reactions are particularly appealing and could be applied to a number of other reaction systems. We report here, for the first time, the synthesis of secondary amides via the carbonylative cross-coupling of arylhalides with benzylamine using carbon monoxide gas (Scheme 1) in a glass fabricated microstructured reactor.
000 m2 m−3 whereas conventional laboratory apparatus does not usually exceed 1000 m2 m−3. A consequence of this increase in specific surface is the enhancement of mass and heat transport in the system. Microreactors specifically designed for multiphase gas–liquid and gas–liquid–solid reactions have been used to carry out a range of reactions, including oxidation,2 fluorination,3 nitration4 and hydrogenation5 reactions, all of which have shown enhanced rates, yields and/or selectivities, compared to their corresponding batch reactions. These improvements are attributed to the increased interfacial contact area generated between the phases within the reactor. The advantages of using microreactors for gas–liquid reactions are particularly appealing and could be applied to a number of other reaction systems. We report here, for the first time, the synthesis of secondary amides via the carbonylative cross-coupling of arylhalides with benzylamine using carbon monoxide gas (Scheme 1) in a glass fabricated microstructured reactor.
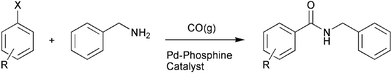 | ||
| Scheme 1 The synthesis of N-benzylbenzamides via carbonylative cross-coupling. | ||
The insertion of carbon monoxide into an organic molecule (known as carbonylation),6 is a convenient but underdeveloped synthetic route to a number of common functional groups including amides, esters, lactams and lactones.7 Carbonylation reactions are usually carried out at high pressures using carbon monoxide gas in the presence of a palladium–phosphine catalyst and can take many hours to reach completion. The rates of these reactions are dependent on a number of factors such as the arylhalide species, the nature of the catalyst, the nucleophilic species, the pressure and the temperature of the system. The insertion step of the carbonylative catalytic cycle (Scheme 2) can be rate limiting due to the poor transport of carbon monoxide into the solution phase. This step of the catalytic cycle can be enhanced by using high pressure reactors, however, such reactors are inherently expensive and require special safety precautions. Microreactor devices provide an unexplored and potentially very useful alternative method for carrying out these gas–liquid phase carbonylation reactions.
 | ||
| Scheme 2 Catalytic cycle for the carbonylation reaction. | ||
Our microfluidic device was constructed from a glass substrate using chemical wet etching techniques.† The liquid and gaseous reagents are mixed on the chip using a mixing-tee configuration. The reaction channel is 5 m long and has an average width of 200 µm and average depth of 75 µm, giving a total volumetric capacity of 75 µl. The area taken up by the microchannel footprint on the glass substrate is approximately 50 mm × 50 mm (Fig. 1).
 | ||
| Fig. 1 A photograph of the microfluidic chip used in the carbonylation reactions prior to chromium removal and bonding. | ||
The liquid reagents, consisting of a mixture of arylhalide, palladium–phosphine catalyst and benzylamine, were infused into the chip at rates of 20, 10 or 5 µl min−1 while the gas flow was kept constant at 2 sccm.‡ An annular flow regime is imposed on the system at these flow rates whereby a thin film of liquid is forced to the surface walls of the microchannel while the gas flows through the centre, this generates a very high interfacial area. This flow pattern was found to be the easiest to reproduce; other flow patterns, such as slug flow, were difficult to control over the length of experimental timescale. Three different arylhalide substrates (iodobenzene 1a, 4-iodoanisole 1b and 2-bromopyridine 1c) were used to test the microreaction carbonylation procedure; the resulting yields are shown in Table 1.
The mean residence time of the microreactions was estimated to be less than 2 min for all the flow rates, based on the timing of fluorescent dyes through the chip.§ The length of the chip device (5 m) is necessary to give the reagents enough time within the channels of the device to react due to the gas forcing the liquid out of the chip, initial experiments using devices with shorter channels (< 2 m) showed only trace amounts of product. Batch reactions were carried out over a ten minute time period to assess the performance of the microreactor.¶ The microreactions, even with their much shorter residence times, show an increase in yield when compared to the batch reactions. When the liquid infusion rates are relatively high (20 µl min−1) modest yields of these conventionally slow reactions are obtained in a very short time period. Reduction of the flow rate to 5 µl min−1 leads to a slight gain in observed product yield. We believe this to be due to a more even distribution of the liquid reagents on the chip's walls forming a more stable annular flow regime.
The increase in yield of the microreactions can be attributed to two factors, the increased interfacial gas–liquid contact area generated within the microstructured reactor and an increase in carbon monoxide pressure resulting from the backpressure produced in the system. The initial carbon monoxide backpressure in the system varied between 3 and 4 bar, depending on the liquid infusion rate. Both of these factors will increase the carbon monoxide solubility and therefore benefit the insertion step of the catalytic cycle.
An interesting side product from the reaction is the α-ketoamide derivative (Fig. 2), formed by a double carbonylation mechanism.8 The α-ketoamide formation was observed for substrates 1a and 1b but not for 1c. Surprisingly, the α-ketoamide was the main product formed by reaction of 1b. No α-ketoamide formation was observed in the batch reactions. As the reaction was not optimised for the formation of the α-ketoamide product it may be possible to enhance its formation by tailoring the reaction conditions through varying the amount of catalyst and the gas/liquid infusion rates.
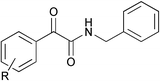 | ||
| Fig. 2 α-Ketoamide product, resulting from the double carbonylation reaction. | ||
Yields obtained for the carbonylation of substrate 1b are lower than those of 1a and 1c. This is due to the introduction of an electron donating methoxy group on the para postion of the phenyl ring. Electron donating groups on arylhalides are generally known to slow down the oxidative addition step of the catalytic cycle and therefore give lower yields. The formation of the α-ketoamide as the major product for the reaction of 1b is due to an enhanced CO insertion rate relative to nucleophilic attack by base as a result of the decreased electrophilicity of the CO-coordinated aryl–palladium complex.8c In contrast, substate 1c shows exclusive formation of the amide, no formation of the α-ketoamide product is observed due to the electron withdrawing nature of the pyridine ring. The gas chromatograms were run after acidic work-up and extraction, therefore any product (N-aryl amine) from a competitive Buchwald-type amination reaction would have been removed in the acidic layer and not be observed on the GC trace. However, unreacted arylhalide was observed in the GC traces, and as suggested by one of the referees a second pass through the reactor or a longer channel should improve overall yields. We intend to investigate further the competitive nature of this type of amination reaction.
In summary, for the first time a microstructured device has been used to perform a gas–liquid carbonylation reaction. The total yields of products obtained are good considering the very short residence times (< 2 min) for these conventionally slow reactions. When compared to traditional batch scale reactions, the gains in yields over this time period obtained by the microreactor are significant and demonstrate a distinct advantage in using such methods. Space–time yields (mol l−1 h−1) achieved by the microreactions are over an order of magnitude greater than the batch reactions. Improvements in yields for these reactions could be achieved by optimising the reaction conditions and catalysts. Specialist uses of these microreaction systems could be applied to catalyst screening9 or in rapid radiolabelling techniques,10 such as applied in Positron Emission Tomography, where the focus is centred on fast chemical syntheses.
Notes and references
- (a) K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406 CrossRef; (b) W. Ehrfeld, V. Hessel and H. Löwe, Microreactors: New Technology for Modern Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
- N. de Mas, R. J. Jackman, M. A. Schmidt and K. F. Jensen, in IMRET 5: Proceedings of the 5th International Conference on Microreaction Technology, ed. M. Matloz, W. Ehrfeld and J. P. Baselt, Springer, Berlin, 2001, p. 60 Search PubMed.
- (a) R. D. Chambers and R. C. H. Spink, Chem. Commun., 1999, 883 RSC; (b) R. D. Chambers, D. Holling and R. C. H. Spink, Lab Chip, 2001, 1, 132 RSC; (c) K. Jähnisch, M. Baerns, V. Hessel, W. Ehrfeld, W. Haverkamp, H. Löwe, C. Wille and A. Guber, J. Fluorine Chem., 2000, 105, 117 CrossRef CAS.
- J. Antes, T. Türcke, J. Kerth, E. Marioth, F. Schnürer, H. H. Krause and S. Löbbecke, in IMRET 5: Proceedings of the 5th International Conference on Microreaction Technology, ed. M. Matloz, W. Ehrfeld and J. P. Baselt, Springer, Berlin, 2001, p. 446 Search PubMed.
- (a) J. Kobayashi, Y. Mori, K. Okamoto, R. Akiyama, M. Ueno, T. Kitamori and S. Kobayashi, Science, 2004, 304, 1305 CrossRef CAS; (b) N. Yoswathananont, K. Nitta, Y. Nishiuchi and M. Sato, Chem. Commun., 2005, 1, 40 RSC.
- (a) H. M. Colquhoun, D. J. Thompson and M. V. Twigg, Carbonylation Direct Synthesis of Carbonyl Compounds, Plenum Press, New York, 1991 Search PubMed; (b) J. Tsuji, Palladium Reagents and Catalysis, Wiley, Chichester, 1995, p. 188 Search PubMed; (c) R. F. Heck, Palladium Reagents in Organic Synthesis, Academic Press, New York, 1985, p. 341 Search PubMed.
- (a) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327 CrossRef CAS; (b) A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318 CrossRef CAS; (c) M. Mori, K. Chiba and Y. Ban, J. Org. Chem., 1978, 43, 1684 CrossRef CAS; (d) A. Cowell and J. K. Stille, J. Am. Chem. Soc., 1980, 102, 4193 CrossRef CAS; (e) L. D. Martin and J. K. Stille, J. Org. Chem., 1982, 47, 3630 CrossRef CAS.
- (a) H. des Abbayes and J. Y. Salaün, Dalton Trans., 2003, 1041 RSC; (b) A. Yamamoto, F. Ozawa, K. Osakada, L. Huang, T. Son, N. Kawasaki and M. K. Doh, Pure Appl. Chem., 1991, 63, 687 CrossRef CAS; (c) F. Ozawa, H. Soyama, H. Yangihara, I. Aoyama, H. Takino, K. Izawa, T. Yamamoto and A. Yamamoto, J. Am. Chem. Soc., 1985, 107, 3235 CrossRef CAS.
- T. Herweck, S. Hardt, V. Hessel, H. Löwe, C. Hofmann, F. Weise, T. Dietrich and A. Freitag, in IMRET 5: Proceedings of the 5th International Conference on Microreaction Technology, ed. M. Matloz, W. Ehrfeld and J. P. Baselt, Springer, Berlin, 2001, p. 215 Search PubMed.
- (a) F. Karimi and B. Långström, J. Chem. Soc., Perkin Trans. 1, 2002, 2111 RSC; (b) H. Audrain, L. Martarello, A. Gee and D. Bender, Chem. Commun., 2004, 558 RSC.
- Z. Guo, E. D. Dowdy, W. S. Li, W. Polniaszek and E. Delaney, Tetrahedron Lett., 2001, 42, 1843 CrossRef CAS.
Footnotes |
| † A soda lime glass substrate with a positive photoresist and a low reflective chromium layer was exposed using a direct-write laser lithographic system (DWL 2.0, Heidelberg Instruments, Heidelberg, Germany) to transfer the channel design. The photoresist was chemically developed and the channels of the design etched into the glass using HF–NH4F solution. External holes were drilled at the reagent entry and exit points. The substrate was then cleaned to remove the excess chromium, soaked in concentrated sulfuric acid and rinsed with deionised water prior to thermal bonding of the glass cover plate. |
| ‡ In a typical reaction a (1 M) solution of arylhalide (0.5 mmol) in benzylamine (0.5 ml) and palladium catalyst (Pd(dppp)Cl2, 2 mol%) was infused into a microfluidic chip at flow rates of 20, 10 or 5 µl min−1 where it was mixed with a steady stream of CO gas, metered at 2 sccm using a Sierra 100 series Smart-Trak mass flow controller. The chip was heated to 80 °C using a heating block. The solution was collected into a vial containing HCl (1 M) to quench the reaction and extracted using dichloromethane (2 × 5 ml). Quantitative analysis was done by gas chromatography. Reference amide materials were synthesised using a similar procedure to Schoenberg and Heck,7a reference α-ketoamide materials were synthesised by a procedure similar to that used by Guo et al.11 |
| § Chip residence times were estimated by injecting a 20 µl slug of fluorescein into a continuous flow of water at infusion rates of 20, 10 and 5 µl min−1 at gas (air) flow rates of 2 sccm. The fluorescent dye was timed from its initial entry onto the chip until its exit; at all infusion rates residence times were less than two minutes. |
| ¶ To a Schlenk flask was added a (1 M) solution of arylhalide (0.5 mmol) in benzylamine (0.5 ml) and palladium catalyst (Pd(dppp)Cl2, 2 mol%). The flask was then placed under an atmosphere of carbon monoxide (filled and evacuated three times). The reaction flask was then placed into a preheated oil bath (80 °C) and stirred vigorously for 10 min. After this time the carbon monoxide atmosphere was removed and the reaction worked-up and analysed in the same way as for the microreaction. |
| This journal is © The Royal Society of Chemistry 2006 |