Lithocholic acid analogues, new and potent α-2,3-sialyltransferase inhibitors†
Kai-Hsuan
Chang
a,
Lenselot
Lee
b,
Jessica
Chen
a and
Wen-Shan
Li
*a
aInstitute of Chemistry, Academia Sinica, Taipei 115, Taiwan. E-mail: wenshan@chem.sinica.edu.tw; Fax: +886(2)2783 1237; Tel: +886(2)2789 8662
bDepartment of Chemistry, National Taiwan Normal University, Taipei 116, Taiwan. Fax: +886(2)2932 4249; Tel: +886(2)2935 0749
First published on 9th January 2006
Abstract
A new type of noncompetitive α-2,3-sialyltransferase inhibitor has been synthesized; we report the discovery, preparation and inhibitory activity of sixteen lithocholic acid analogues.
Sialic acids, nine-carbon amino sugars bearing a negative charge under physiological conditions, are cell-type specific markers that are widely distributed throughout the organism.1 Sialyltransferases (ST), a class of glycosyltransferases, catalyze the transfer of sialic acids to the terminal positions of growing oligosaccharide chains of glycoconjugates.2 Hypersialylation at the non-reducing end of glycoproteins or glycolipids is a vital biological process, which plays a role in various physiological and pathological events, such as cell–cell adhesion, immune defense, tumor cell metastasis, invasions, and inflammation.3 Therefore, the discovery of potent inhibitors of ST has become a promising strategy for chemotherapeutic treatment of cancer4 and can be an invaluable tool to study ST-dependent physiology in immune responses.5 Lately, ST inhibitors with a structural mimetic of transition-state analogues, bisubstrate analogues, donor analogues, and acceptor analogues have been developed based on cytidine monophosphate-N-acetylneuraminic acid (CMP-Neu5Ac) or disaccharides.6 Although these compounds have been demonstrated to effectively inhibit STs, the inhibitors display poor permeability across cell membranes and their clinical applications for studying the biology of glycoconjugates are relatively limited.7 To our knowledge, few ST inhibitors with a cell-permeable property have been reported so far.7–8 Here we describe the discovery and synthesis of lithocholic acid analogues, which employ a steroidal moiety to improve their uptake,9 and demonstrate noncompetitive inhibition of α-2,3-sialyltransferase (α-2,3-ST) in the presence of CMP-Neu5Ac.
Soyasaponin I (1),8 a rigid pentacyclic system with trisaccharide from soybean saponin, was a new type of ST inhibitor and showed a significant inhibition (Ki = 2.1 µM) toward α-2,3-ST in vivo. Compound 1, where the pentacyclic ring is similar to the main skeleton of steroidal compounds (Fig. 1), exhibits important properties relating not only to cell permeability but also to invasive/metastatic behaviors of cancer cell lines.10 Attractive attention was stimulated by this report and we reasoned that some steroid-related compounds might have potential for inhibiting α-2,3-ST activity. As a result of our initial screening efforts, we have identified epiandrosterone succinyl ester (2), a potent Schistosoma japonicum glutathione S-transferase inhibitor,11 and lithocholic acid (3), a substrate of nuclear pregnane X receptor,12 as potent inhibitors of α-2,3-ST with IC50 values of 350 and 21 µM, respectively.
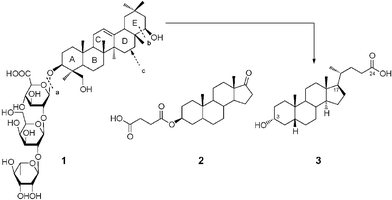 | ||
| Fig. 1 Main skeleton of lithocholic acid 3 is depicted as being formed from 1 by cleaving bonds a and b, followed by converting the D-ring into a 5-membered ring, and oxidizing the terminal alcohol into carboxylic acid. | ||
Synthesis of epiandrosterone derivatives 4–5 and 27–28 was achieved as depicted in Scheme 1. Epiandrosterone 23 was esterified with the required amino acids to yield the corresponding esters 24–26, which were subsequently converted to the epiandrosterone–amino acid conjugates 4 and 27–28 by alkaline hydrolysis of the Fmoc and acidolytic cleavage of the remaining tBu or Boc groups. Efficient formation of epiandrosterone–phosphate conjugate 5 was obtained from 23via a three-step procedure (Scheme 1). The alcohol 23 was converted to the bis-(p-nitrophenyl)epiandrosteronyl phosphate intermediate 29 by carefully controlling the release of one equiv. p-nitrophenol with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), followed by alkaline-mediated replacement of the p-nitrophenyl with methyl. The final product 5 was accomplished by quantitative silyldemethylation of the P,P-dimethyl groups of 30 with bromotrimethylsilane.13 Coupling of succinic anhydride to androsterone in the presence of 4-(dimethylamino)pyridine (DMAP) and pyridine produced the desired isomer 6.
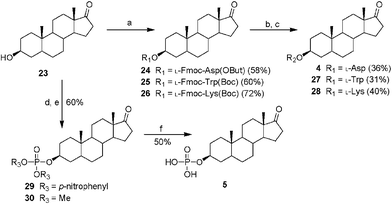 | ||
| Scheme 1 (a) Protected amino acid, DCC, DMAP, DCM, r.t., 30 min; (b) DBU, DCM, r.t., 30 min; (c) trifluoroacetic acid (TFA), 2% H2O or 4 N HCl, MeOH, r.t., 1 h; (d) P(O)(p-nitrophenyl)3, DBU, DCM, r.t., 15 h; (e) DBU, MeOH, DCM, r.t., 15 h; (f) bromotrimethylsilane, DCM, r.t., 30 min. | ||
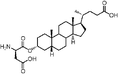
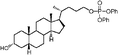
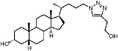
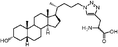
In addition, Scheme 2 illustrates the synthesis of lithocholic acid analogues 7–22. Compound 3 was treated with Amberlite IR120 in anhydrous methanol, and reduction of the resulting methyl ester gave the alcohol 7. The hydroxyl group of 7 was converted to a diphenylphosphate group of 8 in the presence of diphenyl phosphorylazide (DPPA) and DBU. Condensation of 3 and the corresponding protected amino acids, H-Gly-OBut and L-H-Asp(OBut)-OBut, using 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU)/diisopropylethylamine (DIPEA) as coupling agent furnished the protected conjugates 32–33. Compounds 9–10 were obtained after removal of the remaining tBu groups. Analogues 15–16 were prepared by the esterification of 32–33 and succinic anhydride in a manner similar to 6. Oxidation of 3 with CrO3 in acidic conditions produced 11 in 70% yield. Compound 12 was synthesized in a manner similar to that described for 6, 15 and 16 by esterification of 3 with succinic anhydride. In order to prepare analogues 13–14 and 17, the hydroxyl group of 3 was transformed to an acetyl group, and subsequent esterification gave 34. Intermediate 35, derived from 34 by deacylation, was converted to the desired product 13 by dicyclohexylcarbodiimide (DCC)-promoted coupling followed by deprotection of the Fmoc, Boc and tBu groups. Esterification of the secondary alcohol 35 gave compound 14. The D-form amino acid–lithocholic acid conjugate 17 was synthesized in a manner similar to that described for 13. The 1,2,3-triazoles 18–22 required first the synthesis of the terminal azide 31 (Scheme 2). The conversion of diphenylphosphate 8 to the corresponding azide 31 was achieved in the presence of excess sodium azide (NaN3), a catalytic amount of tetrabutylammonium iodide (TBAI), and 15-crown-5.14 Reliable preparation of various 1,4-disubstituted 1,2,3-triazoles 18, 21–22 and 36–37 was successful using the copper(I)-catalyzed ligation of azide 31 and their respective alkynes according to the click chemistry concept approach.17 Subsequent Boc deprotection or saponification afforded the racemic derivatives 19 or 20.
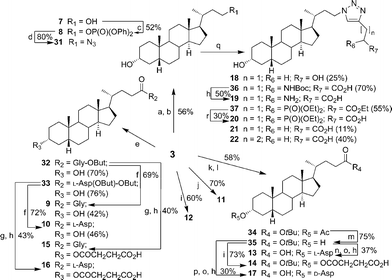 | ||
| Scheme 2 (a) MeOH, Amberlite IR120, 80 °C, 12 h; (b) LAH, THF, 0 °C → r.t., 10 min; (c) DPPA, DBU, THF, r.t., 10 h; (d)NaN3, TBAI, 15-crown-5, 110 °C, 15 h; (e) HBTU, DIPEA, H-Gly-OBut·HCl or L-Asp, N,N-dimethylformamide (DMF), r.t., 30 min; (f) TFA, DCM, r.t., 1 h; (g) succinic anhydride, DMAP, pyridine, 80 °C, 15 h; (h) TFA, 2% H2O, r.t., 1 h; (i) succinic anhydride, DMAP, pyridine, 60 °C, 15 h; (j) CrO3, acetic acid, 100 °C, 30 min; (k) acetic anhydride, pyridine, r.t., 5 h; (l) t-BuOH, DMAP, DCC, DCM, r.t., 30 min; (m) NaOCH3, MeOH, r.t., 2 h; (n) Fmoc-L-Asp(OBut)-OH, DMAP, DCC, DCM, r.t., 30 min; (o) DBU, DCM, r.t., 1 h; (p) Fmoc-D-Asp(OBut)-OH, DMAP, DCC, DCM, r.t., 30 min; (q) alkyne, CuSO4·5H2O, sodium ascorbate, THF/H2O (1 ∶ 1), r.t., 8 h; (r) NaOH, EtOH/H2O (1 ∶ 1), r.t., 5 h. | ||
The inhibition assay of α-2,3-ST was done as described by Schmidt15 using CMP-Neu5Ac as a substrate and the modified disaccharide16 with a 4,5-dimethoxy-2-nitrobenzyl group as a UV-labelled acceptor (Scheme 3). To enhance the potency in compound 2, we initially prepared four derivatives 4–5 and 27–28 with alternations of the succinyl ester unit. However, efforts to improve the IC50 were unsuccessful, for example, replacement of the succinyl moiety of 2 with aspartyl, phosphate, Tyr, or Lys either reduced inhibitory activity (IC50 > 350 µM for 4–5) or gave a product displaying no biological functions (27–28) toward α-2,3-ST (Table 1). In addition, a minor influence on the inhibitory property (IC50 = 450 µM for 6) of the α isomer of 2 has been observed, suggesting negligible interaction in the recognition of epiandrosterone/androsterone analogues with succinyl-stereochemistry by the enzyme.
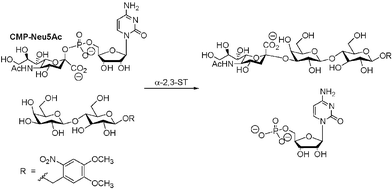 | ||
| Scheme 3 α-2,3-ST catalyzed sialylation of acceptor. | ||
| Compound | # | IC50 (µM)b | Compound | # | IC50 (µM)b |
|---|---|---|---|---|---|
| a Note that rat liver α-2,3-ST was obtained from CalBiochem at a concentration of 1 mU/µL. b The inhibition assay was carried out as described in ref. 15, except for the use of the modified disaccharide16 as the acceptor. | |||||
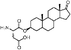
|
4 | 480 |

|
14 | 13 |

|
5 | 400 |
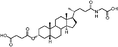
|
15 | 22 |

|
6 | 450 |
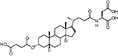
|
16 | 18 |
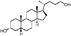
|
7 | 351 |
|
17 | 6 |
|
8 | > 100 |
|
18 | > 100 |

|
9 | 25 |
|
19 | 83 |

|
10 | 16 |
|
20 | 7 |
|
11 | 139 |

|
21 | 10 |
|
12 | 12 |
|
22 | 5 |
|
13 | 12 | |||
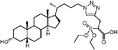
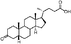
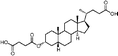
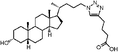
Lithocholic acid (3), which potentially mimics a pentacyclic ring of 1 (Fig. 1), showed a promising inhibition constant (IC50 = 21 µM), indicating an acceptable pharmacophore. Among the 16 synthetic analogues (Table 1), the terminal alcohol 7 and its derivative 8 displayed a 5–17-fold decrease over 3, suggesting that a carboxylic acid group is important for promoting affinity. This was further confirmed by utilizing the method of peptide coupling to extend the terminal carboxylic acid; the inhibitory properties of compounds 9 and 10 can be restored completely compared to those of 7–8. To determine the importance of the 3-hydroxyl position of 3, the inhibition of α-2,3-ST activity by compound 11, which has a ketone moiety in place of a hydroxyl group, was evaluated. Compound 11 was found to have an IC50 of 139 µM, representing 7-fold lower potency than 3. Furthermore, compounds 12 and 13 exhibited a 2-fold potency increase over 3, indicating that construction of the 3-hydroxyl portion of 3 is tunable to the interaction of binding affinity. When the terminal carboxylic acid of 12 was varied, the analogues 14–16 appeared to reach a low micromolar affinity plateau (Table 1). Surprisingly, replacement of the D-Asp with L-Asp, as in 17, gave a 2-fold improvement in potency over 13. In addition, compound 3 was further optimized by replacing the carboxylic acid with a 1,4-disubstituted 1,2,3-triazole ligand using the click chemistry approach. Compounds 20 and 21 showed an 8–12-fold potency increase over 19, suggesting that adding the positively charged amino group could abolish affinity. The primary alcohol 18 was at least 10-fold less potent than 21 and 20-fold less potent than 22. These results are consistent with previous observations concerning the value of the terminal carboxylic acid in binding. Analogue 22 was the most active compound with an IC50 of 5 µM. The Lineweaver–Burk plot (Fig. 2) shows that compound 22 exhibits a noncompetitive inhibition (Ki = 2.2 µM) toward cytidine monophosphate N-acetylneuraminic acid (CMP-Neu5Ac). However, we failed to identify the mode of inhibition between 22 and acceptor due to the solubility problem derived from the modified disaccharide with a 4,5-dimethoxy-2-nitrobenzyl moiety. It is possible that compound 22 binds to α-2,3-ST and alters the structure of the active site, resulting in the decrease of enzyme activity. Due to the potential of compounds 11, 13, 15–16, and 19–22 as anticancer drugs, our collaborators recently evaluated their cytotoxicity using in vitro assays against several tumor cell lines. These results will be published elsewhere.
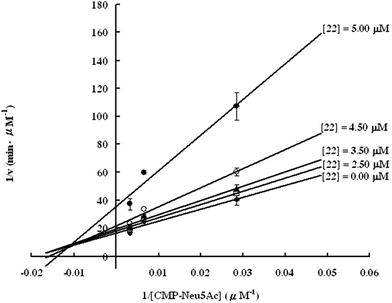 | ||
| Fig. 2 Lineweaver–Burk plot of the results of rat α-2,3-ST inhibition assay for the synthetic inhibitor 22. | ||
Because of the physiological and pathological significance of ST, there is strong demand for an efficient approach to searching for cell-permeable inhibitors. This is the first report of a strategy for designing effective α-2,3-ST inhibitors based on the structure of an authentic inhibitor. Analogues 17, 20, and 22 display more potent inhibitory activities among these synthetic derivatives. The results demonstrated here should pave the way for the design of other ST inhibitors.
We are grateful for funding from the National Science Council (NSC 94-2113-M-001-017) and Academia Sinica (program project).
Notes and references
- C. Traving and R. Schauer, Cell. Mol. Life Sci., 1998, 54, 1330 CrossRef CAS; M. Schwarzkopf, K.-P. Knobeloch, E. Rohde, S. Hinderlich, N. Wiechens, L. Lucka, I. Horak, W. Reutter and R. Horstkorte, Proc. Natl. Acad. Sci. USA, 2002, 99, 5267 CrossRef CAS.
- A. Harduin-Lepers, V. Vallejo-Ruiz, M.-A. Krzewinski-Recchi, B. Samyn-Petit, S. Julien and P. Delannoy, Biochimie, 2001, 83, 727 CrossRef CAS.
- E. C. Seales, G. A. Jurado, B. A. Brunson, J. K. Wakefield, A. R. Frost and S. L. Bellis, Cancer Res., 2005, 65, 4645 CrossRef CAS; R. Kannagi, M. Izawa, T. Koike, K. Miyazaki and N. Kimura, Cancer Sci., 2004, 95, 377 Search PubMed; L. K. Mahal, N. W. Charter, K. Angata, M. Fukuda, D. E. Koshland and C. R. Bertozzi, Science, 2001, 294, 380 CrossRef CAS.
- M. M. Fuster and J. D. Esko, Nat. Rev. Cancer, 2005, 57, 526 CrossRef.
- M. A. Daniels, K. A. Hogquist and S. C. Jameson, Nat. Immunol., 2002, 3, 903 CrossRef CAS.
- D. Skropeta, R. Schwoerer, T. Haag and R. R. Schmidt, Glycoconjugate J., 2004, 21, 205 CrossRef CAS; K.-H. Chang, Y. S. Tao and W.-S. Li, Synlett, 2004, 37 CAS; L. J. Whalen, K. A. McEvoy and R. L. Halcomb, Bioorg. Med. Chem. Lett., 2003, 13, 301 CrossRef CAS; R. Schwoerer and R. R. Schmidt, J. Am. Chem. Soc., 2002, 124, 1632 CrossRef CAS; B. Muller, C. Schaub and R. R. Schmidt, Angew. Chem., Int. Ed., 1998, 37, 2893 CrossRef CAS.
- K.-Y. Lee, H. G. Kim, M. R. Hwang, J. I. Chae, J. M. Yang, Y. C. Lee, Y. K. Choo, Y. I. Lee, S.-S. Lee and S.-I. Do, J. Biol. Chem., 2002, 277, 49341 CrossRef CAS; A. K. Sarkar, T. A. Fritz, W. H. Taylor and J. D. Esko, Proc. Natl. Acad. Sci. USA, 1995, 92, 3323 CAS.
- T.-W. Lin, W.-W. Chang, C.-C. Chen and Y.-C. Tsai, Biochem. Biophys. Res. Commun., 2005, 331, 953 CrossRef CAS; C.-Y. Wu, C.-C. Hsu, S.-T. Chen and Y.-C. Tsai, Biochem. Biophys. Res. Commun., 2001, 284, 466 CrossRef CAS.
- P. Yu, B. Liu and T. Kodadek, Nat. Biotechnol., 2005, 23, 746 CrossRef CAS.
- C. C. Hsu, T. W. Lin, W. W. Chang, C. Y. Wu, W. H. Lo, P. H. Wang and Y. C. Tsai, Gynecol. Oncol., 2005, 96, 415 CrossRef CAS.
- S.-C. Jao, J. Chen, K. Yang and W.-S. Li, Bioorg. Med. Chem., 2006, 14, 304 CrossRef CAS.
- S. J. Kim, E.-K. Bang, H. J. Kwon, J. S. Shim and B. H. Kim, ChemBioChem, 2004, 5, 1517 CrossRef CAS.
- H. B. Wood and B. Ganem, Tetrahedron Lett., 1993, 34, 1403 CrossRef CAS.
- F. Liu and D. J. Austin, J. Org. Chem., 2001, 66, 8643 CrossRef CAS.
- C. Schaub, B. Müller and R. R. Schmidt, Glycoconjugate J., 1998, 15, 345 CrossRef CAS.
- S. B. Cohen and R. L. Halcomb, J. Org. Chem., 2000, 65, 6145 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: α-2,3-ST assay, general experimental procedures and compound characterization. See DOI: 10.1039/b514915k |
| This journal is © The Royal Society of Chemistry 2006 |