DNA-duplexes containing abasic sites: correlation between thermostability and acoustic wave properties
T.
Hianik
a,
X.
Wang
b,
S.
Andreev
c,
N.
Dolinnaya
c,
T.
Oretskaya
c and
M.
Thompson
*b
aLaboratory of Biophysics, Faculty of Mathematics, Physics and Computer Science, Comenius University, Mlynská dolina F1, 842 48 Bratislava, Slovak Republic
bDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario, Canada M5S 3H6. E-mail: mikethom@chem.utoornto.ca
cChemistry Department and A.N. Belozersky Institute of Physico-Chemical Biology, M.V. Lomonosov Moscow State University, Leninskije Gory, Moscow, 119 992, Russia
First published on 15th August 2006
Abstract
Aldehydic apurinic or apyrimidinic sites that lack a nucleobase moiety are one of the most common forms of toxic lesions in DNA. In the present study, a close structural analog of such a site, the 2-(hydroxymethyl) tetrahydrofuranyl residue, was synthesized in order to act as a model for damaged nucleic acid probes. Prepared oligodeoxyribonucleotides containing one, two or three abasic sites were hybridized to complementary sequences immobilized on a gold surface using the neutravidin–biotin interaction for study by thickness shear mode acoustic wave detector. Measurement of the complex electrical impedance of an AT-cut quartz device with immobilized biotinylated nucleotide allowed the detection of changes of series resonance frequency, Δfs, and motional resistance, Rm, associated with duplex formation. The changes as detected by the acoustic wave method correlated well with the thermostability of DNA duplexes in solution. With respect to the latter, UV-monitored melting curves indicate that both the number of sites and their localization in the double-stranded structure influence the amount by which a 19 b.p. duplex is destabilized. The presence of 3 abasic sites completely destabilized the DNA duplex.
Introduction
DNA damage can arise from various routes, including oxidative stress, the action of environmental pollutants, e.g. nitrates or organophosphate pesticides, and attack by the high-energy radioactive process.1 Base damage, such as the spontaneous deamination of cytosine to uracil, the oxidation of thymine to thymine glycol or the oxidation of guanine can be repaired via abasic site intermediates. The first step in base excision repair of DNA containing damaged bases in vivo is often the hydrolytic cleavage of the C–N bond between the sugar and damaged or abnormal base to generate an aldehydic apurinic or apyrimidinic site (AP). Such sites constitute one of the most common forms of toxic lesion. They are generated both enzymatically during the repair of DNA damage and spontaneously, as a result of chemical modification of the bases (by carcinogens, alkylating agents or by ionization radiation) that destabilizes N-glycosidic bonds.2 The unpaired aldehydic abasic site changes the structural and dynamic properties of DNA that affect the recognition and misreading of lesions by the cellular machinery involved in DNA repair and replication. The presence of these lesions has been shown to slow down, but not block RNA polymerases, virus reverse transcriptase,3 and can cause mis-incorporation of nucleoside triphosphates by a DNA polymerase during replication.4,5 Thus, the changes in nucleotide sequences of DNA, when replicated and transmitted to future cell generations, become the source of permanent mutations. Accumulation of mutations is connected directly with aging and cancer. In view of the biological significance of AP sites, there is considerable interest in the detection of such lesions.Three-dimensional structures of DNA duplexes containing native and synthetic abasic sites have been determined by NMR spectroscopy.6,7 In these studies, the duplex retained the B-form geometry and the AP site induced only local perturbations limited to the lesion site and flanking base pairs. In duplexes containing purine residues opposite the lesion, both the AP site and opposing purine are stacked inside the helix. However, when pyrimidine bases are opposite to the AP-site, the duplex structure shows increased conformational flexibility. Depending on the temperature and the nature of nucleobases flanking AP sites, the abasic sugar residue and, occasionally, the opposite pyrimidine bases are extrahelical.8 Since AP sites change base stacking and hydrogen bonding, they can also affect the thermodynamic properties of the DNA duplex.9 Accordingly, the presence of AP sites can also affect DNA hybridization. Thus, the study of DNA hybridization between single-stranded oligonucleotide (probe) and damaged (target) oligonucleotide can serve as a test of AP-site damage. For this purpose the model probe oligonucleotides (ODNs) are chemically modified by thiol groups through short linker or by biotinylation in order to effect attachment to the device by means of chemisorption or by avidin–biotin technology.10 Hybridization with damaged target ODN can be detected at the electrode surface by a number of electrochemical methods,11,12 by means of scanning Kelvin nanoprobe,13 acoustic wave14–16 or optical methods.17,18 The study of the mechanisms of DNA hybridization using naturally occurring AP sites is difficult, because they are unstable. This is due to the fact that the aldehyde (open) form is subject to β-elimination and strand scission.19 This limitation can be overcome by using the 2-(hydroxymethyl)tetrahydrofuran-3-ol residue, a close structural analog of a natural abasic site. We synthesized the series of 19-mer oligodeoxyribonucleotides (ODN) containing one, two or three tetrahydrofuran AP analogs at the desired positions (AP1, AP2 and AP3 respectively), where hybridization with a complementary strand (BASE) incorporates abasic sites opposite T residues. This allows the creation of a model where the type of damage and the distance separating the lesions are known. Unmodified oligomer APC was used as a control. In order to immobilize the DNA on a gold electrode we used a biotinylated 19-mer BASE probe. The probe ODNs were immobilized to a gold support by means of neutravidin.20 The detection of DNA hybridization has been performed by means of the acoustic wave propagation technique using the thickness shear mode device (TSM). This method is based on measurement of the complex impedance of the high frequency oscillation of an AT-cut quartz piezoelectric sensor. The TSM method has been shown to be a powerful tool for the study of binding events involving biochemical macromolecules at the liquid–device interface.21
Additionally, a solution-based study of the thermal stability of the identical DNA duplexes containing AP sites was performed in order to conduct a correlation between acoustic wave properties and the UV-monitored melting behavior of DNA duplexes.
Experimental
Reagents
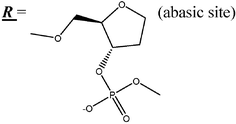
19-mer ODNs with one, two and three AP-sites (Table 1) were synthesized by the phosphoramidite method on an automated DNA synthesizer Applied Biosystems 380 B. The modified residues were introduced by replacement of standard nucleoside phosphoramidite with 2-(dimethoxytrityloxymethyl)tetrahydrofuran-3-ol (N,N-diisopropylamido)-β-cyanoethylphosphite under the standard conditions of oligonucleotide synthesis. The ODN modified by biotin (X-BASE, Table 1) was purchased from Thermo Electron Corporation (Germany). The oligonucleotide concentration was determined spectrophotometrically with UV-VIS Hitachi 150-20 (Japan) or UV1700 Shimadzu (Japan) spectrophotometers.![[X with combining low line]](https://www.rsc.org/images/entities/b_char_0058_0332.gif)
Preparation of DNA layers on the TSM electrode
Biotinylated ODNs were immobilized to only one side of an AT-cut quartz crystal with a gold electrode in place. The device was first carefully cleaned in acetone, and then ethanol followed by methanol in an ultrasound sonicator. It was then washed in deionized water and dried in a stream of nitrogen. After cleaning, the crystal was placed into a flow-through cell. The gold electrode of the sensor was flushed with buffer A solution (0.5 M NaCl, 10 mM Tris-HCl, 1 mM EDTA, pH 7.6) continuously for 30 min and then treated by neutravidin as follows. Neutravidin (Pierce, Rockford, IL, USA) dissolved in DPBS buffer (Sigma, St. Louis, USA) to a concentration of 1 mg ml−1 was allowed to flow into the cell at a flow rate 62.5 µl min−1. After approx. 10 min the signal reached a steady-state value. Then buffer A was allowed to flow at the same rate again for 10 min. Subsequently, X-BASE at a concentration of 1 µM dissolved in buffer A was pumped over the device surface. Buffer A was flowed for a further 10 min to wash away any non-specific binding probes. Finally, ODNs with or without abasic sites dissolved in buffer A to a concentration of 1 µM were swept over the device for the same time period and buffer A was allowed to wash away the non-specific binding of ODNs.Acoustic wave detection of abasic sites in oligonucleotide DNA duplexes
The thickness-shear mode acoustic method is a highly sensitive tool for the on-line detection of biochemical binding events at surfaces. It is based on application of an electric field to an AT-cut quartz crystal through electrodes which results in the generation of an acoustic shear wave in the bulk crystal. The sensing layer is placed at one side of the crystal. The shear wave propagates through the film and into the liquid and can be characterized from storage and dissipation processes. The measured electrical impedance of the sensor is related to the complex acoustic impedance. As a result the series resonance frequency fs and motional resistance Rm are determined. The value of fs represents the energy storage and reflects the mass changes of the oscillating layer, while Rm is related to energy dissipation and provides evidence of changes of shearing viscosity of the layer.22 In order to study DNA hybridization by means of the TSM method, we used 9 MHz AT-cut piezoelectric crystals with gold electrodes (International Crystals Manufacturing Co Inc., Oklahoma City, OK, USA). The crystal was incorporated into a flow-through system as previously described in detail.23 Only one side of the crystal was exposed to liquid flow in a flow cell, while the second side was exposed to nitrogen gas. A network analyzer (HP4395A Network/Spectrum analyzer, Hewlett Packard, Colorado Springs, Co, USA) was used to measure the impedance properties of the sensor. The sensing layer has been formed as described above.UV-monitored thermal denaturation of DNA duplexes
Absorption spectra and thermal denaturation profiles were measured with a Varian-Cary 50 Bio UV-VIS spectrophotometer (USA) equipped with a thermoelectrically controlled cell holder. For melting experiments, the absorbance of samples at 260 nm was measured between 20 and 80 °C every 2 degrees. The samples containing 2 µM DNA duplexes BASE-AP1, BASE-AP2, BASE-AP3 (Table 2) were prepared by mixing in 1 : 1 molar ratio of AP-containing strands with BASE in a buffer B (10 mM Tris-HCl, 10 mM MgCl2, 100 µg ml−1 BSA, 1 mM DTT, pH 7.6). Fully matched duplex BASE-APC was used as a control. The samples were heated to 80 °C, cooled slowly and incubated at room temperature 10 min prior to melting studies.![[R with combining low line]](https://www.rsc.org/images/entities/b_i_char_0052_0332.gif) )
)
| DNA duplex | Designation | Type of helix-coil transition | T m a ± 1 °C | ΔTmb |
|---|---|---|---|---|
| 5′ → 3′ | ||||
| 3′ → 5′ | ||||
| a T m—melting temperature. b ΔTm—the difference in melting temperature for perfect control and modified duplexes. | ||||
| GGAAAATTTCAGCAAGGTG | BASE-APC | Monophasic | 64 | — |
| CCTTTTAAAGTCGTTCCAC | ||||
| GGAAAATTTCGCAAGGTG |
BASE -AP1 | Biphasic | 55 | 9 |
| CCTTTTAAAGTCGTTCCAC | 41 | 23 | ||
| GGAAAATTTCGCAGGTG |
BASE -AP2 | Monophasic | 45 | 19 |
| CCTTTTAAAGTCGTTCCAC | ||||
| GGAAATTTCGCAGGTG |
BASE -AP3 | No cooperative transition | — | — |
| CCTTTTAAAGTCGTTCCAC | ||||
Results and discussion
Study of duplex formation by acoustic wave propagation
The acoustic wave technique was employed for study of the mechanism of hybridization of oligodeoxyribonucleotides containing 1, 2 or 3 abasic sites by comparison with undamaged complementary and non-complementary ODNs. Fig. 1 shows the changes in fs and Rm, following attachment to the device surface of the various compounds. The addition of neutravidin in DPBS results in a sharp decrease of the resonance frequency and increase of the Rm value. The motional resistance is characterized by complex behavior. After an initial increase, immediately following addition of neutravidin, the Rm value reached a maximum and then decreased. Addition of buffer A results in a further decrease of frequency and increase of Rm. After approx. 30 min the changes of both fs and Rm stabilized. The changes of frequency due to addition of neutravidin were approximately 200 Hz, while Rm only slightly decreased in comparison with the initial value, prior to addition of neutravidin. The decrease of series resonance frequency following addition of neutravidin is a well-known phenomenon,23 and is connected with attachment to a gold surface through hydrophobic interactions. According to the Sauerbrey24 expression, from changes in frequency (Δfs) it is possible to produce a rough idea of the change of mass at the device surface, Δm, via the known molecular mass of neutravidin (60 kD). Accordingly, it is potentially feasible to arrive at an estimate of the number of neutravidin molecules chemisorbed at the gold surface of the electrode. According to Sauerbrey, the resonance frequency of the crystal linearly depends on its mass:| Δfs = −2.26 × 10−6f02(Δm/A), |
 | ||
| Fig. 1 Changes of series resonance frequency Δfs (full line) and motional resistance Rm (dashed line) following addition of neutravidin (1), biotinylated DNA (X-BASE, see Table 1) (2) and complementary ODN (APC, see Table 1) (3). (B) represents flow of buffer A. The points of addition are indicated by arrows. | ||
From Fig. 1 it is seen that the flow of buffer A results in a further decrease of the frequency together with a change of Rm value. This effect often accompanies cases where there is an alteration of ionic strength and electrolyte composition. After the frequency and Rm values were stabilized, the biotinylated 19-mer ODN (biotin is placed at the 5′ terminal of ODN) dissolved in buffer A in a concentration of 1 µM was allowed to flow into the cell. Due to the strong affinity of biotin to neutravidin, a strong binding of biotinylated ODN to the neutravidin molecules takes place. This results in a decrease of frequency and increase of Rm value. Flow of buffer results in a slight decrease of the frequency and in a considerable decrease of Rm. The steady-state value of Rm is, however, similar to that of the crystal covered by the neutravidin layer prior to addition of the biotinylated ODN. Probably, the high flexibility of single-stranded DNA does not contribute substantially to the changes of surface viscosity of the layer. On the other hand, considerable changes of Rm following binding of ODN and a slow relaxation of Rm to a steady state value may offer an indication of a substantial structural change in the ODN layer. From the change of the frequency it is possible to estimate the number of ODN molecules adsorbed at a neutravidin layer. As it is evident from Fig. 1, the binding of biotinylated ODN to a neutravidin layer is accompanied by a decrease of the frequency of 60 Hz. Since the molecular weight of ODN is 6300 D, the number of ODN molecules at the surface would be 4.4 × 1012. Thus, according to the Sauerbrey treatment, the number of attached ODNs is about 2.9 times higher than the number of neutravidin molecules chemisorbed at the crystal surface. Radiochemical labeling experiments have shown previously that this ratio of nucleic acid to protein is actually around 1.223 This result again serves notice that the Sauerbrey mass-response model can at best be regarded to yield only approximate values. Other factors in the liquid control acoustic wave propagation.
Injection of complementary ODN (concentration 1 µM) results in a further decrease of the resonance frequency (Fig. 1). The changes of frequency are approx. 52 Hz, which is slightly lower than the changes of the frequency that are typical for adsorption of biotinylated ODN. The differences between these values are, however, not significant. The changes of frequency following addition of complementary ODN are obviously connected with hybridization at the crystal surface. In contrast, the motional resistance increases following addition of complementary ODN. Even after flow of the buffer, the Rm value is higher in comparison with that prior to addition of complementary ODN. These changes are evidence that the double-stranded DNA array at the surface contributes considerably to the increase of surface viscosity. This phenomenon can be explained in term of lower flexibility of double-stranded (dsDNA) in comparison with single-stranded ODN. Certainly, the persistent length of dsDNA is approx. 50 nm.26 The length of 19 b.p. DNA is approx. 6 nm, which is less than the persistent length. Therefore ODNs are in an extended conformation or declined and, therefore, cause more viscous forces between the ODN layer and surrounding electrolyte.
Fig. 2 shows a control experiment after addition of neutravidin and the non-complementary ODN. The result is that there are no changes of frequency and Rm caused by this addition. The non-complementary ODN also did not change the frequency or Rm after addition to the surface with immobilized biotinylated probe (X-BASE). Further buffer flow removes all non-complementary ODN in the cell. Then the ODN containing one abasic site was added at a concentration of 1 µM. This addition results in a decrease of frequency and increase of Rm. These changes are, however, lower in comparison with that caused by hybridization of complementary ODN. Addition of ODN containing 2 abasic sites causes similar changes to that presented in Fig. 1. Presence of three abasic sites in ODN, however, changes the situation dramatically (Fig. 3). As seen from Fig. 3, addition of ODN with 3 abasic sites results in a decrease of frequency, with the flow of buffer A yielding an increase of frequency. The resulting frequency changes are considerably lower in comparison than that for ODNs with one or two abasic sites (see Table 3). This result presents evidence regarding the weak interaction of this ODN with the biotinylated ODN probe at the surface of the device. The changes of frequency and resistance for ODN containing different numbers of abasic sites are summarized in Table 3.
 | ||
| Fig. 2 Changes of series resonance frequency Δfs (full line) and motional resistance Rm (dashed line) following addition of neutravidin (1), non-complementary ODN (BASE, see Table 1) (2), biotinylated DNA (X-BASE, see Table 1) (3), non-complementary ODN (BASE) (4) and ODN contained one abasic site (AP1, see Table 1) (5). (B) represents flow of buffer A. The points of addition are indicated by arrows. | ||
 | ||
| Fig. 3 Changes of series resonance frequency Δfs (full line) and motional resistance Rm (dashed line) following addition of neutravidin (1), biotinylated DNA (X-BASE) (2) and ODN contained three abasic sites (AP3, see Table 1) (3). (B) represents flow of buffer A. The points of addition are indicated by arrows. | ||
| ODN | Δfs/Hz | R m/Ohm |
|---|---|---|
| APC | 49.0 ± 2.9 | 1.7 ± 0.5 |
| AP1 | 39.6 ± 4.5 | 1.3 ± 0.5 |
| AP2 | 42.0 ± 7.4 | 0.7 ± 0.5 |
| AP3 | 13.7 ± 7.0 | −0.3 ± 0.5 |
| BASE | 0 | 0 |
Thermal stability of oligonucleotide duplex DNAs containing abasic sites from UV-detection
19-mer DNA duplexes differing in the number of AP sites (0–3) were characterized by their melting behavior in buffer B (Table 2 and Fig. 4). In the case of the fully paired duplex BASE-APC, the UV-monitored absorption as a function of temperature showed a single reversible transition with a melting temperature (Tm) = 64 ± 1 °C; Fig. 4A demonstrates the melting curves presented in integral and differential forms. Inspection of UV-melting data reveals that the presence of AP sites leads to significant duplex destabilization. This effect depends dramatically on the quantity of AP sites and their location in the double helix. In contrast to the non-modified double helix, BASE-AP1 containing one abasic residue demonstrates the biphasic melting profile reflecting the superposition of two helix-coil transitions. This result corresponds to denaturation of two duplex domains, the boundary being at the AP site, which induces DNA-bending and increased conformational flexibility. Because the AP site is not located in the middle of double helix, we can separately observe the transitions corresponding to short and long duplex domains (with Tm = 41 and 55 °C, respectively, Table 2). In contrast, the incorporation of an abasic site opposite to the deoxyadenosine residue stacked inside the helix does not necessarily alter the melting cooperativity of the short DNA duplex relative to the fully paired Watson–Crick structure.26 Additionally, the second AP site placed in the short domain (BASE-AP2) decreases the Tm of the corresponding transition (Fig. 4C). Therefore, a conversion of the biphasic melting profile to the monophasic type takes place. It should be noted that the Tm of BASE-AP2 (45 °C) containing two AP sites does not equal the dissociation temperature (55 °C) for a long domain of BASE-AP1. The reduced Tm can be attributed to a destabilizing effect of the unpaired 9-nt “tail”. A third AP site placed at the center of the long domain prevents stable duplex formation. The absence of a cooperative S-shaped melting curve points to an unstable structure (Fig. 4D). | ||
| Fig. 4 UV melting curves measured at 260 nm (in integral (1) and differential (2) forms) for 19 b.p. DNA duplexes with zero, one and two abasic sites: BASE-APC (A), BASE-AP1 (B) and BASE-AP2 (C), respectively. The superposition of the melting curves for undamaged duplex BASE-APC (○) and duplex containing three abasic sites BASE-AP3 (•) (D). All measurements were performed in buffer B at a single strand concentration of 4 µM. | ||
These data indicate the significant role of local structural perturbations at AP sites on duplex thermostability. Furthermore, this work confirms that at the temperature conventionally used in electrochemical experiments (20 ± 1 °C), the regular 19-mer ODNs and DNA molecules with one or two abasic sites are in duplex form with a complementary strand present. In contrast, ODN with three AP residues is predominantly in the single-stranded state.
Conclusions
The influence of AP sites on local base stacking energy and geometry causes a dramatic destabilization of the DNA duplex structure. These effects correlate with changes of acoustic wave properties as determined by change of series resonance frequency, and motional resistance. The high sensitivity of the acoustic shear wave technique to the interaction of DNA strands with damaged DNA containing AP sites provides a useful tool for the detection of DNA damage generated by organophosphate pesticides or other agents causing DNA depurination.Acknowledgements
The authors are grateful for support provided by the NATO SfP Program (Project No. SfP – 978003), the Slovak Grant Agency (Project No. 1/1015/04) and by the Natural Sciences and Engineering Research Council of Canada.References
- B. Alberts, D. Bray, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Essential Cell Biology, Garland Publishing, Inc, New York, 1998 Search PubMed.
- L. Loeb and B. Preston, Annu. Rev. Genet., 1986, 20, 201 CrossRef CAS.
- M. Takeshita, C.-N. Chang, F. Johnson, S. Will and A. P. Grollman, J. Biol. Chem., 1987, 262, 10171 CAS.
- L. A. Loeb and T. A. Kunkel, Annu. Rev. Biochem., 1982, 51, 429 CrossRef CAS.
- B. Weiss and L. Grossman, Adv. Enzymol. Relat. Areas Mol. Biol., 1987, 60, 1 Search PubMed.
- R. D. Beger and P. H. Bolton, J. Biol. Chem., 1998, 273, 15565 CrossRef.
- Y. Coppel, N. Berthet, C. Coulombeau, C. Coulobeau, J. Garcia and J. Lhomme, Biochemistry, 1997, 36, 4817 CrossRef CAS.
- Ph. Cuniasse, G. V. Fazakerly, W. Guschlbauer, B. Kaplan and L. C. Sowers, J. Mol. Biol., 1990, 213, 303 CAS.
- A. C. Gelfand, G. E. Plum, A. P. Grollman, F. Johnson and K. J. Breslauer, Biochemistry, 1998, 37, 7321 CrossRef CAS.
- M. I. Pividori, A. Merkoci and S. Alegret, Biosens. Bioelectron., 2000, 15, 291 CrossRef CAS.
- E. Palecek and M. Fojta, Anal. Chem., 2001, 73, 74A CAS.
- J. Wang, Anal. Chim. Acta, 2003, 500, 247 CrossRef CAS.
- M. Thompson, L.-E. Cheran, M. Zhang, M. Chacko, H. Huo and S. Sadeghi, Biosens. Bioelectron., 2005, 20, 1471 CrossRef CAS.
- S. Tombelli, M. Mascini, C. Sacco and A. P. F. Turner, Anal. Chim. Acta, 2000, 418, 1 CrossRef CAS.
- X. C. Zhou, L. Q. Huang and S. F. Y. Li, Biosens. Bioelectron., 2001, 16, 85 CrossRef CAS.
- H. Su and M. Thompson, Biosens. Bioelectron., 1995, 10, 329 CrossRef CAS.
- P. Piunno, U. J. Krull, R. Hudson, M. J. Damha and H. Cohen, Anal. Chem., 1995, 67, 2635 CrossRef CAS.
- X. Chen, X.-E. Zhang, Y.-Q. Chai, W.-P. Hu, Z.-P. Zhang, X.-M. Zhang and A. E. G. Cass, Biosens. Bioelectron., 1998, 3–4, 451 CrossRef.
- A. E. Sudina, E. M. Volkov, T. S. Oretskaia, S. Kh. Degtiarev, D. A. Gonchar and E. A. Kubareva, Bioorg. Khim. (Mosk), 2000, 26, 442 Search PubMed.
- B. A. Cavic and M. Thompson, Anal. Chim. Acta, 2002, 469, 101 CrossRef CAS.
- B. A. Cavic, G. L. Hayward and M. Thompson, Analyst, 1999, 124, 1405 RSC.
- J. S. Ellis and M. Thompson, Phys. Chem. Chem. Phys., 2004, 6, 4928 RSC.
- M. Tassew and M. Thompson, Anal. Chem., 2002, 74, 5313 CrossRef CAS.
- G. Sauerbrey, Z. Phys., 1959, 155, 206 CAS.
- X. C. Zhou, L. Q. Huang and S. F. Y. Li, Biosens. Bioelectron., 2001, 16, 85 CrossRef CAS.
- S. May, D. Harries and A. Ben-Shaul, Biophys. J., 2000, 78, 1681 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |