Multidimensional HRMAS NMR: a platform for in vivo studies using intact bacterial cells
Wei
Li
Department of Pharmaceutical Sciences, College of Pharmacy, University of Tennessee Health Science Center, 847 Monroe Avenue, room 327A, Memphis, TN 38163, USA. E-mail: wli@utmem.edu; Fax: +1-901-448-6828; Tel: +1-901-448-7532
First published on 12th June 2006
Abstract
In vivo analysis in whole cell bacteria, especially the native tertiary structures of the bacterial cell wall, remains an unconquered frontier. The current understanding of bacterial cell wall structures has been based on destructive analysis of individual components. These in vitro results may not faithfully reflect the native structural and conformational information. Multidimensional High Resolution Magic Angle Spinning NMR (HRMAS NMR) has evolved to be a powerful technique in a variety of in vivo studies, including live bacterial cells. Existing studies of HRMAS NMR in bacteria, technical consideration of its successful application, and current limitations in studying true human pathogens are briefly reviewed in this report.
Bacterial cell walls play critical roles in their survival in harsh environmental conditions, pathology, and resistance development to existing drugs. In addition, many of the structural components in the bacterial cell wall do not exist in humans, making the enzymes involved in cell wall biosynthesis fertile targets for anti-bacterial drug development. As with all living systems, bacterial cell walls have a very complicated structure, consisting of integrated macromolecules such as carbohydrates, lipids, and proteins. This structure is highly heterogeneous among individual bacterial cells, due to constant biosynthesis, assembly, disassembly, and turnover.1 Despite decades of extensive research, the understanding of bacterial cell wall structure is far from clear. Controversial cell wall models have been proposed in the literature, with experimental evidence seeming to support one or the other.1,2
There are many challenges to the understanding of bacterial cell wall structure and functions. These include the clearly defined pathways in cell wall biosynthesis, the interactions of key components (lipids, carbohydrates and proteins) in the native cell wall structure, and ultimately, the true three dimensional structure of the cell wall. The major obstacles in bacterial cell wall structure elucidation are their complexity and the lack of a faithful representation of pure compounds in vitro. Traditionally, cell wall structure analyses are performed in a destructive manner, consisting of degradation, isolation, purification and subsequent analysis using various techniques. While these in vitro techniques are extremely fruitful and indispensable for structural elucidation, they are not without limitations. First, these processes are time consuming, labor intensive and normally require substantial quantities of materials. For slow growing bacteria this becomes very expensive or even impossible. Second, due to potential changes induced by chemical isolation, stability after isolation, and variability among different isolates, there is no guarantee that the isolated structure will faithfully retain the native identity, relative abundance, and in vivo structure.3–5 The final complications are that these degradative analyses may result in loss of information about the native conformation and dynamics. The loss or alteration of the conformational information may mask critical details on their ability to perform in vivo functions. The isolation of each individual component, while greatly facilitating the elucidation of a particular structure, largely destroys potential intermolecular interactions. Therefore, analytical methods that provide in vivo information could yield important and novel insights into cell wall structures. One promising analytical method is the recently developed high resolution magic angle spinning NMR (HRMAS NMR) technique which is the focus of the current review. Specifically, we focus on recent applications of multidimensional 1H and 13C HRMAS NMR to study intact bacterial cells.
Initially developed in the late 1990s, HRMAS NMR can be considered a hybrid technique between solid state NMR and classical solution state NMR.6 Similar to solid state NMR, the use of magic angle spinning (MAS) effectively removes spectral line broadening resulting from chemical shift anisotropy, homonuclear dipolar interactions, and magnetic susceptibility.7,8 These line broadening effects are reduced to zero because the (3 cos2θ − 1)/2 part of the Hamiltonian disappears when the sample is spinning along the magic angle of θ = 54.7° with respect to the static magnetic field (B0). This technology produces narrow lines in heterogeneous samples such as tissues or whole cells.9–14 At the same time it retains the deuterium locking and low power levels in classical solution NMR experiments. Similar to solution NMR, HRMAS NMR involves direct polarization transfer and not cross polarization transfer (CPMAS) between 1H and heteronuclei (13C or 15N), thus distinguishing it from CPMAS experiments on true solids. A distinct advantage of this technique is that HRMAS NMR rotors typically contain less than 100 µl of sample and spin at the magic angle. Additionally, the rotor spinning rate is relatively low (up to a few kHz) thus maintaining the integrity of intact cells while removing the significant spinning side bands from interested spectral regions.
Intact cells consist of molecules that differ dramatically in their abundance, size, location and relative mobility. The inherent low sensitivity of NMR will only detect abundant and mobile components. Therefore, for 1D proton HRMAS NMR, a rotor-synchronized Carr–Purcell–Meibom–Gill (CPMG) pulse sequence coupled with water suppression is normally used. This sequence generates a better baseline by removing “chemical backgrounds”, resulting from superimposition of molecules in low abundance and/or with restricted mobility.15 The ability to provide a narrow spectral line width using very small amounts of heterogeneous samples has prompted the application of HRMAS NMR techniques to in vivo bacterial studies in the last few years.16–20
Using 1H and 13C HRMAS NMR and only 1 mg of sample, Jachymek and co-workers characterized and made a full assignment of the O-specific polysaccharides in isolated lipopolysaccharides (LPS) from four strains of Yokenella regensburgei.18 These O-polysaccharide components were also directly observed in their original form on the surface of living bacterial cells using HRMAS NMR. This demonstrates the potential to use HRMAS NMR as a fast screening or quality control method without an extensive isolation/purification process. Szymanski and co-workers recently examined capsular polysaccharides directly in intact Campylobacter cells using HRMAS NMR. They demonstrated the feasibility of using this technique for serotying, mutant verification, and preliminary sugar analysis in Campylobacter jejuni and Neisseria meningitidis.19,21 The ability to use HRMAS NMR on live bacterial cells to differentiate the relative composition of the bacterial cell wall, to demonstrate targets for anti-bacterial drugs, and to verify gene mutation were further demonstrated by Lee and co-workers (Fig. 1).17 They studied 2D 1H–13C HSQC (heteronuclear single quantum correlation) HRMAS NMR of untreated Mycobacterium smegmatis intact cells (Fig. 1a, control), cells treated with a known anti-mycobacterium drug ethambutol (EMB) (Fig. 1b), and cells having embB mutation (Fig. 1c). Each peak in the anomeric region (1H: 4.8 ppm to 5.5 ppm; 13C: 90 ppm to 110 ppm) in these 2D spectra corresponds to an anomeric CH pair of a sugar unit in the cell wall structure. The substantial reduction or absence of a peak in the spectra indicates that the corresponding biosynthetic pathway responsible for that sugar unit in the cell wall is inhibited. When comparing the HRMAS NMR spectra of EMB treated and control Mycobacterium smegmatis intact cells, they directly observed the inhibiting effects of EMB on arabinosyltransferases (EmbA–C), an enzyme for cell wall biosynthesis (the absence of the circled peak in Fig. 1b). A subsequent HSQC spectrum using an embB knockout mutant in M. smegmatis clearly validated this inhibition effect (the substantial reduction of the circled peak in Fig. 1c). In another study, Szymanski and co-workers tracked the biosynthesis of N-linked glycans in live Campylobater jejuni using 1H HRMAS NMR and they were able to quantify the glycosylation efficiency within the live cells.22
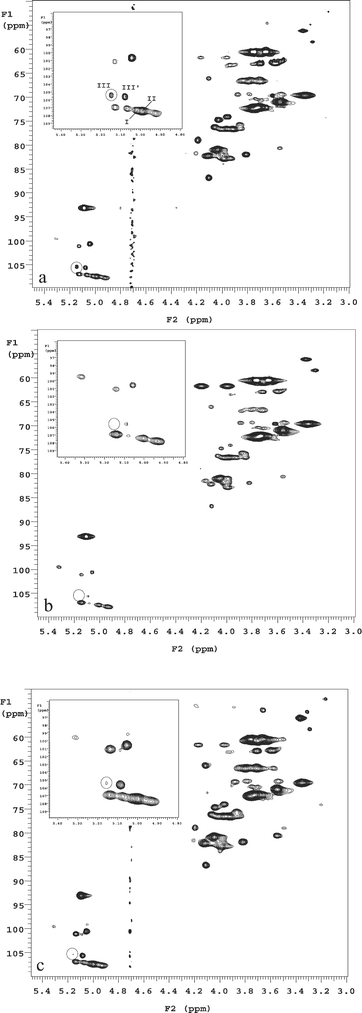 | ||
| Fig. 1 HRMAS 1H–13C HSQC NMR spectra of whole cell M. smegmatis (a) untreated control, (b) treated with 50 µg ml−1 EMB for 6 h, and (c) M. smegmatis ΔembB mutant. Circled regions denote the cross-peak from 2-α-Araf (III) residue in the cell wall. Roman numerals correspond to the arabinan residues: I, 3,5-α-Araf; II, 5-α-Araf; III, 2-α-Araf→3; III′, 2-α-Araf→5. Reproduced with permission from ref. 17. | ||
The potential for using HRMAS NMR as a non-invasive tool to monitor drug metabolism in vivo is also exciting as recently demonstrated by Lippens and co-workers.20 Using 1D 1H and 2D 1H–1H TOCSY (total correlation spectroscopy) HRMAS NMR, they were able to detect a novel metabolite produced in live mycobacteria from a pro-drug at a therapeutically relevant concentration as low as 5 µg ml−1. Combined with diffusion filtering, the results from HRMAS NMR indicated that this metabolite is located inside the intact cells which has not been detected by any previous in vitro methods. This study was done with unlabeled precursor molecule and at a relatively low proton frequency of 300 MHz, demonstrating the great potential of HRMAS NMR for in vivo structural analysis as well as drug metabolism studies.
While 1D NMR is fast and can provide important information, the limited resolution and severe peak overlaps obtained from whole bacterial cells are often far from satisfactory. Multidimensional NMR possesses enormous potential in whole cell NMR. Many sets of 2D, 3D, or high dimensional solution NMR techniques exist and are the primary tools in NMR based structural biology. 23 However, the complex nature of the pulse sequences, the unique geometry of the rotor, and the fast rotation of the samples along the magic angle, require special consideration for successful application of multidimensional HRMAS NMR using whole cells.24 Straightforward applications of solution state multidimensional NMR sequences may give poor signal/noise ratios in the spectrum, or even fail completely.25,26 Proper experimental conditions for successful application of multidimensional HRMAS NMR using whole cell bacteria need to be worked out.
In a recent report, Li and co-workers examined several parameters for observing cell wall signals using Mycobacteria smegmatis.26 They systematically examined the effect of rotor synchronization on different pulse sequences, acquisition time, isotope labeling strategies, and the effect of temperature. Proper rotor synchronization was shown to be critical for multidimensional HRMAS NMR using bacterial cells in which many pulses and gradients are involved. Imperfections in rotor synchronization can be rapidly amplified by the large numbers of pulses and gradients, resulting in reduced signal intensity. For example, when the pulses and gradients were optimized for a spin rate of 2 kHz in a 3D 1H–13C HCCH–COSY experiment, rotor spinning at 1.5 kHz resulted in a nearly complete signal loss while spinning at 2.0 kHz showed very strong signals. Another critical factor is to develop suitable isotope labeling strategies. Except for 1H–1H homonuclear 2D NMR, stable isotope labeling is preferred in whole cell NMR for less abundant nuclei (13C or 15N). In fact, many of the 3D NMR relies on the magnetization transfer between proton and heteroatom, and it is impractical to run these experiments without enrichment due to the low natural abundance of these nuclei. Proper isotope labeling can not only greatly enhance the sensitivity of the signals but also provide important selectivity in different biological pathways using whole cells.26–31 It is also advantageous to run whole cell NMR at elevated temperature, not only to mimic the physiological conditions (e.g. 37 °C), but also to enhance signal intensity due to increased mobility of molecules. Combining results from multidimensional NMR of whole cells and purified sample standard, this method may provide unique insights into intact bacterial cells, as demonstrated by the example of assigning the t-β-araf sugar unit in a major cell wall carbohydrate, arabinogalactan (AG), in Mycobacterium smegmatis (Fig. 2).26 In this approach, peaks that severely overlapped in the 2D 1H–13C HSQC whole cell spectrum (Fig. 2c) were well resolved by analysis of the 3D whole cell 1H–13C HCCH–COSY spectrum (Fig. 2a and Fig. 2b). Fig. 2a combines all the 13C(F2)–1H(F3) planes from the 3D HCCH–COSY spectrum into one 2D frame to construct a projection of the 3D spectra. This allowed visualization of all the detected spin systems in the whole cell at one time. The assignment of each spin system, using the t-β-araf sugar unit as an example, was done by analyzing the well separated individual planes of the 3D data (Fig. 2b). Subsequently these assignments are confirmed with literature reported assignments from the 2D 1H–13C HSQC spectrum of the purified AG standard (Fig. 2d).
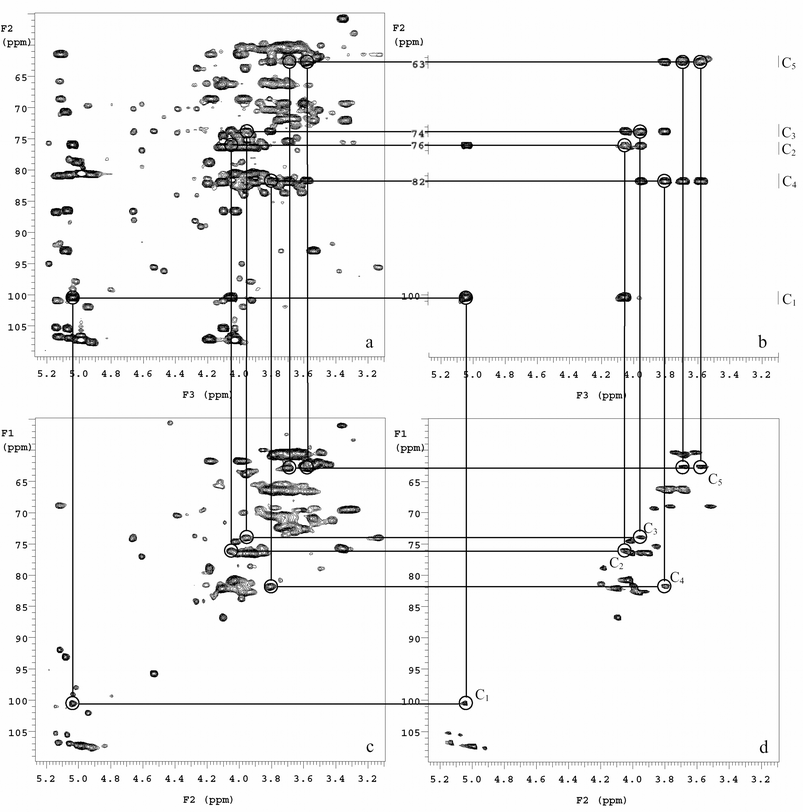 | ||
| Fig. 2 Analysis of the t-β-araf spin system in the cell wall of M. smegmatis using 3D 1H–13C HCCH–COSY and 2D 1H–13C HSQC HRMAS NMR. (a) Projection of the 3D data in the 13C(F2)–1H(F3) plane allowing visualization of all spin systems acquired in the whole-cell M. smegmatis spectrum. The anomeric cross-peak for the t-β-araf was defined in the 3D projection (circled) and subsequently identified along with the cosy δH shift pattern in the individual 3D planes. (b) Partial sections of the five separate 13C(F2)–1H(F3) planes, which shows the correlation among C1–C5 of t-β-araf. The carbon and proton shifts were compared to (c) the 2D 1H–13C HSQC of M. smegmatis whole cells and (d) the 2D 1H–13C HSQC spectrum of base solubilized AG standard. Reproduced with permission from ref. 26 | ||
The potential applications of various HRMAS NMR techniques using intact bacterial cells, in combination with existing chemical and biological tools, are very exciting. One of these potential applications will be the development of proper labeling schemes and parameter optimization to study the nuclear Overhauser effect (NOE) in bacterial cell wall structures. Intramolecular NOEs can provide information about the native conformation, while the observation of inter-macromolecular NOEs will provide exciting direct information to study and refine current models of cell wall structures. Due to the severe spectral overlap using 2D 1H–1H NMR (NOESY),18 3D NMR methods such as 1H–13C NOESY–HSQC may be the best choice for such studies.
The use of multidimensional HRMAS NMR to study intact bacterial cells has its limitations. While one can obtain a decent 2D 1H–13C HSQC spectrum in 15 minutes using a well labeled sample, an acceptable 3D 1H–13C HCCH–COSY experiment will take a few hours. Other 3D NMR such as 1H–13C HCCH–TOCSY or NOESY–HSQC requires higher resolution therefore it typically takes 24 hours or longer to acquire the data. NMR rotors are tightly sealed so there is no added oxygen or nutrition. There is no doubt that cells will have biochemical responses during lengthy HRMAS NMR experiments. However, it has been shown that the bacterial survival rate after two hours of NMR measurement is higher than 93%, and there are no detectable changes in the 1H–13C HSQC spectra before and after 24 hours of 3D HRMAS NMR measurements.26 The current NMR techniques usually detect major components in the bacterial cell wall, and these components are unlikely to change substantially in this time period. At this stage, 2D 1H–13C HRMAS NMR seems to be the most robust for whole cell studies, while 3D HRMAS NMR is reserved to facilitate peak assignment for 2D NMR. Additionally, these methods enable the study of macromolecular structure/interactions which are not substantially altered during NMR experiments. Improving signal sensitivity and selectivity will be the key for wide applications of 3D NMR. With the rapid advances of both NMR hardware (e.g., the cryogenic NMR probe32) and the development of various fast NMR data acquisition methods (e.g., G-matrix Fourier transform,33 projection-reconstruction,34 and covariance NMR35), multidimensional HRMAS NMR has the potential to become an invaluable tool in bacterial research.
Quantification of signals observed in intact cells will be important in areas from drug discovery to gene mechanism studies. One has to be very cautious in solely relying on signal intensity (or peak integral) in HRMAS NMR spectra, as the intensity will be affected by the location and mobility of molecules. Molecules in rigid environments may escape detection even if they are present. Therefore, combination of HRMAS NMR with traditional chemical and biological techniques is highly desirable.
Specific to the pathogenic nature of bacterial cells, developing strict protocols for the safe handling of real human pathogens for HRMAS NMR studies seems to be the biggest obstacle at present.36 While sample preparation and disposal can be performed safely in a hood, there is always a risk of rotor failure at high spinning speed and the possible release of harmful bacteria. So far, all the studies reported in the literature employed relatively safe bacteria. The availability of crash-proof HRMAS rotors and proper incorporation of a biosafety retaining system such as bacterial HEPA filters in the probe are a pre-requisite before any attempt to expand these techniques directly to true human pathogens.
In conclusion, although there are many obstacles to overcome, multidimensional HRMAS NMR using intact bacterial cells represents a novel platform that could provide important in vivo information. Complementary to existing chemical and biological methods, this technique may prove to be an invaluable tool in drug development, gene function validation, and the study of virulence mechanisms.
Acknowledgements
The author wishes to thank the reviewers for their insightful comments and Dr Bob Moore, II for his critical reading of the manuscript. This work is supported by funds from the College of Pharmacy, University of Tennessee Health Science Center.References
- B. Dmitriev, F. Toukach and S. Ehlers, Trends Microbiol., 2005, 13, 569–574 CrossRef CAS.
- W. Vollmer and J. V. Holtje, J. Bacteriol., 2004, 186, 5978–5987 CrossRef CAS.
- D. J. McNally, H. C. Jarrell, J. Li, N. H. Khieu, E. Vinogradov, C. M. Szymanski and J. R. Brisson, FEBS J., 2005, 272, 4407–4422 Search PubMed.
- A. E. DeBarber, K. Mdluli, M. Bosman, L. G. Bekker and C. E. Barry, Proc. Natl. Acad. Sci. USA, 2000, 97, 9677–9682 CrossRef CAS.
- J. Duus, C. H. Gotfredsen and K. Bock, Chem. Rev., 2000, 100, 4589–4614 CrossRef CAS.
- A. E. Manzi, and P. A. Keifer, in Techniques in Glycobiology, ed. R. R. Townsend and A. T. Hotchkiss, Jr., Marcel Dekker, Inc.: New York, NY, 1997, pp. 1–16 Search PubMed.
- E. R. Andrews and R. G. Eades, Nature, 1959, 183, 1802 CAS.
- A. N. Garroway, J. Magn. Reson., 1982, 49, 168–171 CAS.
- L. L. Cheng, M. J. Ma, L. Becerra, T. Ptak, I. Tracey, A. Lackner and R. G. Gonzalez, Proc. Natl. Acad. Sci. USA, 1997, 94, 6408–6413 CrossRef CAS.
- P. Weybright, K. Millis, N. Campbell, D. G. Cory and S. Singer, Magn. Reson. Med., 1998, 39, 337–345 CrossRef CAS.
- M. G. Swanson, D. B. Vigneron, Z. L. Tabatabai, R. G. Males, L. Schmitt, P. R. Carroll, J. K. James, R. E. Hurd and J. Kurhanewicz, Magn. Reson. Med., 2003, 50, 944–954 CrossRef CAS.
- I. F. Duarte, E. G. Stanley, E. Holmes, J. C. Lindon, A. M. Gil, H. Tang, R. Ferdinand, C. G. McKee, J. K. Nicholson, H. Vilca-Melendez, N. Heaton and G. M. Murphy, Anal. Chem., 2005, 77, 5570–5578 CrossRef CAS.
- B. Martinez-Granados, D. Monleon, M. C. Martinez-Bisbal, J. M. Rodrigo, J. del Olmo, P. Lluch, A. Ferrandez, L. Marti-Bonmati and B. Celda, NMR Biomed., 2006, 19, 90–100 CrossRef CAS.
- D. J. Philp, W. A. Bubb and P. W. Kuchel, Magn. Reson. Med., 2004, 51, 441–444 CrossRef CAS.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CAS.
- J. Czaja, W. Jachymek, T. Niedziela, C. Lugowski, E. Aldova and L. Kenne, Eur. J. Biochem., 2000, 267, 1672–1679 CrossRef CAS.
- R. E. B. Lee, W. Li, D. Chatterjee and R. E. Lee, Glycobiology, 2005, 15, 139–151 CAS.
- W. Jachymek, T. Niedziela, C. Petersson, C. Lugowski, J. Czaja and L. Kenne, Biochemistry, 1999, 38, 11788–11795 CrossRef CAS.
- S. K. Gudlavalleti, C. M. Szymanski, H. C. Jarrell and D. S. Stephens, Carbohydr. Res., 2006, 341, 557–562 CrossRef CAS.
- X. Hanoulle, J.-M. Wieruszeski, P. Rousselot-Pailley, I. Landrieu, A. R. Baulard and G. Lippens, Biochem. Biophys. Res. Commun., 2005, 331, 452–458 CrossRef CAS.
- C. M. Szymanski, F. St. Michael, H. C. Jarrell, J. Li, M. Gilbert, S. Larocque, E. Vinogradov and J.-R. Brisson, J. Biol. Chem., 2003, 278, 24509–24520 CrossRef CAS.
- J. Kelly, H. Jarrell, L. Millar, L. Tessier, L. M. Fiori, P. C. Lau, B. Allan and C. M. Szymanski, J. Bacteriol., 2006, 188, 2427–2434 CrossRef CAS.
- J. Cavanagh, W. J. Fairbrother, A. G. Palmer, III, and N. J. Skelton, Protein NMR Spectroscopy: Principles and Practice, Academic Press, Inc., San Diego, CA, 1996 Search PubMed.
- M. Piotto, K. Elbayed, J. M. Wieruszeski and G. Lippens, J. Magn. Reson., 2005, 173, 84–89 CrossRef CAS.
- A. S. Zektzer, M. G. Swanson, S. Jarso, S. J. Nelson, D. B. Vigneron and J. Kurhanewicz, Magn. Reson. Med., 2005, 53, 41–48 CrossRef.
- W. Li, R. E. B. Lee, R. E. Lee and J. Li, Anal. Chem., 2005, 77, 5785–5792 CrossRef CAS.
- Z. Serber, W. Straub, L. Corsini, A. M. Nomura, N. Shimba, C. S. Craik, P. Ortiz de Montellano and V. Doetsch, J. Am. Chem. Soc., 2004, 126, 7119–7125 CrossRef CAS.
- Z. Serber, L. Corsini, F. Durst and V. Doetsch, Methods Enzymol., 2005, 394, 17–41 CrossRef CAS.
- S. Reckel, F. Loehr and V. Doetsch, ChemBioChem, 2005, 6, 1601–1606 CrossRef CAS.
- D. S. Burz, K. Dutta, D. Cowburn and A. Shekhtman, Nat. Methods, 2006, 3, 91–93 CrossRef CAS.
- J. E. Bryant, J. T. J. Lecomte, A. L. Lee, G. B. Young and G. J. Pielak, Biochemistry, 2005, 44, 9275–9279 CrossRef CAS.
- G. Chapdelaine and P. Cleon, Spectra Anal., 2003, 32, 35–38 Search PubMed.
- S. Kim and T. Szyperski, J. Am. Chem. Soc., 2003, 125, 1385–1393 CrossRef CAS.
- E. Kupce and R. Freeman, J. Am. Chem. Soc., 2004, 126, 6429–6440 CrossRef CAS.
- F. Zhang and R. Bruschweiler, J. Am. Chem. Soc., 2004, 126, 13180–13181 CrossRef CAS.
- J. M. Wieruszeski, P. Talaga and G. Lippens, Anal. Biochem., 2005, 338, 20–25 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |