Solid-supported room temperature phosphorescence from aflatoxins for analytical detection of Aspergillus spp. strains
T.
Rojas-Durán
,
I.
Sánchez-Barragán
,
J. M.
Costa-Fernández
and
A.
Sanz-Medel
*
Physical and Analytical Chemistry Department, University of Oviedo, Julian Claveria, 8, Oviedo 33006, Spain. E-mail: asm@uniovi.es; Fax: +34 985103125
First published on 9th May 2006
Abstract
A simple, direct and rapid analytical methodology for the detection of aflatoxin producing Aspergillus spp. strains based on the measurement of room temperature phosphorescence from aflatoxins is presented here.
Moulds belonging to Aspergillus genus are common contaminants of commodities intended for human consumption and animal feed. Some of them have the ability to produce aflatoxins.1,2
Aflatoxins are the mycotoxins most commonly detected in foodstuffs. They have proved to be carcinogenic, mutagenic, hepatotoxic and immunosuppressive. Therefore, development of screening analytical methodologies for the rapid identification of aflatoxigenic strains is a research field of great present interest.
Microbiological methods used for the identification of aflatoxin producing strains consist of culturing isolated Aspergillus strains in agar plates containing different additives that are known to promote the production of aflatoxins. When cultures are observed under UV light, aflatoxigenic colonies emit a blue-green or bright-blue fluorescence not detected when non-aflatoxigenic strains are cultured. Widespread microbiological methods proposed to date require between 3 and 10 days to provide a positive result.3–5
Recently, a method based on the addition of cyclodextrins (CD) plus sodium deoxycholate (NaDC) to the culture media has been reported for the identification of aflatoxigenic strains, through the observation of a beige ring around aflatoxigenic colonies which fluoresces under UV light.6 In further experiments, using this modified culture, a long-lived green luminescence emission was observed after turning off the UV light lamp. Considering that this green-emission persisted for some time (less that one second) after removal of incident radiation, it could be a room temperature phosphorescence emission from chemical species present in the media.
The experimental conditions typically required in order to obtain analytically useful phosphorescence are: absence of oxygen (as it deactivates the triplet excited state of the phosphor); the presence of a heavy atom in order to favor the intersystem crossing rate; and finally, a proper environment (such as rigid supports) in which the phosphor is immobilized.
For the first time, we report in this article that, under some optimized experimental conditions, aflatoxins produced by artificial cultures of Aspergillus flavus emit analytically useful RTP emission (even without adding any deoxygenating reagent, or a heavy atom to the medium) that can be used to detect foodstuff contaminations by those highly toxic strains.
The in vivo RTP emission observed in the presence of oxygen could be explained by the “anti-oxygen-quenching room temperature phosphorescence mechanism” proposed by Li et al.7 In addition, the semi-solid nature of the modified culture medium (increased by the addition of NaDC), hinder the non-radiative deactivation of the triplet excited state and allow RTP to be clearly observed. Furthermore, the addition of an external heavy atom to the medium was not necessary to obtain analytically useful RTP. This was also observed by other authors for other phosphorescent analytes.8
The proposed method for the identification of aflatoxin producing Aspergillus spp. strains adds the analytical advantages provided by phosphorescent detection to the advantages of the inexpensive and simple microbiological methods: wide separation between the excitation and emission spectra and low background emission. Moreover, since phosphorescence is less common than fluorescence, the selectivity of the detection is highly increased. In addition, interferences from any short-lived fluorescence and scattered light can be easily avoided using an appropriate delay time.9
In our experiments, firstly the capability of aflatoxin standard solutions to emit phosphorescence at room temperature was evaluated (such luminescent emission had never been reported before). Experimental and chemical conditions were optimized, first for in vitro conditions, investigating room temperature phosphorescence detection from aflatoxins standard solutions†. The optimized procedure was then applied to measure the RTP emission from standard solutions of aflatoxin B1, B2, G1 and G2, which were all immobilized on an appropriate solid surface, ensuring the presence of a heavy atom perturber (in order to populate the triplet excited state) and oxygen displacement (by means of an argon stream) in order to prevent “quenching” of the RTP signal. Safety cautions were used during handling and disposal of the analytes as they are considered dangerous and highly toxic substances‡.
It was observed that, in such experimental conditions, a highly intense RTP emission from immobilized aflatoxins in silica gel beads (and in the presence of 2-bromoethanol) can emit strong RTP signals centred at about 500 nm and having triplet lifetimes of the luminescent emission of about 500 µs. Fig. 1 and Table 1 summarize the spectroscopic characteristics (emission spectra, RTP lifetimes and wavelengths) of such emissions for the different aflatoxins assayed.
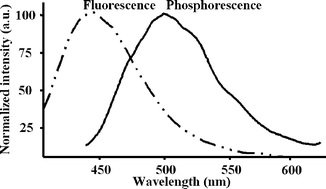 | ||
| Fig. 1 Fluorescence and room temperature phosphorescence emission spectra of aflatoxin B1 immobilized over silica gel beads.|| | ||
| Aflatoxin | Lifetime/µs | Max λex/nm | Max λem/nm |
|---|---|---|---|
| B1 | 512 ± 9 | 377 | 487 |
| B2 | 488 ± 10 | 374 | 485 |
| G1 | 571 ± 10 | 386 | 502 |
| G2 | 547 ± 11 | 387 | 506 |
| Mixture | 527 ± 12 | 370 | 500 |
It can be observed that due to a spectral overlapping of the emission maxima it is not possible to discriminate between the different aflatoxins in a mixture just by using direct RTP emission intensity measurements.
In order to prove the relationship between the production of aflatoxins and the RTP emitted by mould cultures, two representative strains (supplied by the Spanish Type Culture Collection, CECT, Valencia, Spain) were cultured on malt extract agar with CAVASOL® and sodium deoxycholate§: one was non-aflatoxigenic, A. flavus strain (CECT 2685) and the other one was aflatoxigenic A. parasiticus strain (CECT 2681). After 3 days of incubation at 32 °C, the chloroform extracts from these cultures were obtained as described elsewhere9 and aflatoxins immobilized following the procedure described below for the aflatoxin standards.†
The aflatoxigenic strain CECT 2681 produces the four aflatoxins B1, B2, G1 and G2. Since they all have analogous molecular structures, their RTP emission maxima are also very similar to each other (see Table 1). Thus, the spectrum obtained from the extract of the aflatoxigenic strain was similar to that from aflatoxin B1 standard (as shown in Figs. 1 and 2). On the other hand, the RTP spectrum of the extract from the non-aflatoxigenic strain yielded a small RTP emission, but it was negligible compared to the intensity from the aflatoxigenic strain extracts.
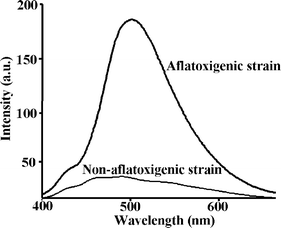 | ||
| Fig. 2 Room-temperature phosphorescence emission spectra of immobilized chloroform extracts from an A. parasiticus CECT 2681 aflatoxigenic culture compared to the chloroform extract from an A. flavus CECT 2685, a non-aflatoxigenic culture. Both extracts were immobilized onto silica-gel beads.† | ||
Once it was confirmed that the observed RTP emission is due to the production of aflatoxins in culture media, a simple test was developed in order to evaluate the performance of RTP detection for “real” sample analysis ¶. Slants containing modified malt extract agar (MACD) §were inoculated with four representative aflatoxigenic (CECT 2680, 2681, 2682 and 2687) and two non-aflatoxigenic strains (CECT 2684 and 2685). The culture plates were then incubated at 32 °C. Each culture was carried out in triplicate. The acquisition of the RTP signal from the cultures was carried out every 3 hours||, measuring directly on the Petri dishes by means of a fiber optic accessory connected to the luminescence spectrometer (see Fig. 3). Laboratory biosafety practices were always followed while these assays were carried out‡.
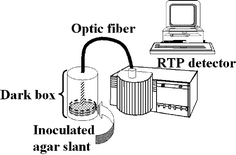 | ||
| Fig. 3 Schematic diagram of the experimental system for in vivo RTP acquisition. | ||
The results obtained demonstrated that aflatoxigenic strains can be positively identified and distinguished from non-aflatoxigenic strains within the first 36 hours of incubation, by means of the measurement of aflatoxins native RTP emission (see Fig. 4). The RTP detection method described here improves significantly those currently in use needing between 3 and 10 days for confirmatory results. Additionally, this proposed RTP methodology is more selective than that using conventional spectrofluorimetric detection (there are many other possible coexisting fluorescent species that could result in false positive results). Moreover, direct fluorescence measurements of the samples were also carried out simultaneously to the RTP measurements, and it turned out that the agar and the micellium of the mould itself show a very intense fluorescence emission. As a result, even after more than 2 days of culture, the fluorescence signal of aflatoxins could not be discriminated from this high background.
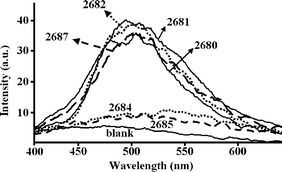 | ||
| Fig. 4 RTP spectra from cultures of aflatoxigenic strains CECT 2680, 2681, 2682 and 2687 versus non-aflatoxigenic strains CECT 2684 and 2685. “Blank” corresponds to the RTP spectrum of a non-inoculated agar slant. | ||
In order to evaluate the actual analytical capability of the proposed test, all the assayed cultures were also analysed by means of a reference alternative HPLC-fluorescence method described elsewhere.10
It was confirmed that chloroform extracts of the aflatoxigenic strain cultures that exhibited RTP signal certainly contained aflatoxins, while extracts from cultures of the negative strain did not.
Conclusions
For the first time, room temperature phosphorescence emission from aflatoxins has been detected in vitro by means of immobilization of these toxic analytes onto an appropriate solid support and by ensuring the presence of a heavy atom.A correlation between the RTP emission observed in vivo and the production of aflatoxins by Aspergillus strains in culture medium has been established.
Results so far have demonstrated the great analytical potential of the proposed RTP methodology for the development of a screening test to identify aflatoxin producing strains which is simpler and faster as compared to classical microbiological techniques previously used for the purpose.
This work was supported by the SWIFT-WFD (from the 6th F.P. of the European Union) and by the project BQU2003-04671 (from the Ministery of Education and Science, Spain). Authors wish to thank Dr Wei Jun Jin from Shanxi University for his suggestions on this work. The modified malt extract agar (MACD§) described in this work is the patented property of the University of Santiago de Compostela. ES 2 195 771. April, 2002.
References
- J. W. Cary, M. A. Klich and S. B. Beltz, Mycologia, 2005, 97, 425–432 Search PubMed.
- J. C. Frisvad, P. Skouboe and R. A. Samson, Syst. Appl. Microbiol., 2005, 28, 442–53 Search PubMed.
- N. D. Davis, S. Iyer and U. L. Diener, Appl. Environ. Microbiol., 1987, 53, 1593–1595 CAS.
- S. K. Dyer and S. McCammon, J. Appl. Bacteriol., 1994, 76, 75–78 CAS.
- P. A. Lemke, N. D. Davis, S. K. Iyer and G. W. Creech, Appl. Environ. Microbiol., 1989, 55, 1808–1810 CAS.
- J. Jaimez Ordaz, C. A. Fente, B. I. Vázquez, C. M. Franco and A. Cepeda, Int. J. Food Microbiol., 2003, 83, 219–225 CrossRef.
- G. R. Li, J. J. Wu, W. J. Jin and J. W. Xie, Talanta, 2003, 60, 555–562 CrossRef CAS.
- W. J. Jin, Y. Wei, A. Xy, C. H. Liu and S. Zangh, Spectrochim. Acta, 1994, 50A(10), 1769–1775 CrossRef.
- A. Sanz-Medel, Anal. Chim. Acta, 1993, 283, 367–378 CrossRef CAS.
- T. R. Rojas, C. A. Fente, B. I. Vázquez, C. M. Franco and A. Cepeda, J. Food Control, 2005, 16, 445–450 CrossRef CAS.
- J. M. Costa-Fernández and A. Sanz-Medel, Anal. Chim. Acta, 2000, 407, 61–69 CrossRef CAS.
Footnotes |
| † According to the method described elsewhere,11 silica gel beads (160 µm particle size, Sigma, USA) were conditioned before analysis by washing with hydrochloric acid solution (50% v/v), water, ethanol solution (50% v/v) and once more with water. The conditioned beads (0.5 g) were spiked out with 200 µL of aflatoxins standard solutions (10−6 M in acetonitrile) and 50 µL pure 2-bromoethanol (Sigma Aldrich, USA). This preparation was allowed to dry overnight inside a stove at 30 °C, then the spiked beads were packed in a conventional flow-through cell (Hellma, Müllheim, Germany). At the bottom of the cell, a small piece of nylon net was placed to prevent particle displacement by the gas stream. The flow cell was placed inside the sample-holder of a Cary Eclipse (Varian Ibérica, Madrid, Spain) luminescence spectrometer equipped with a flash Xe arc source. The luminescence cell was then connected to an argon flow to dry and deoxygenate it for 5 minutes before RTP emissions were acquired. Blank samples were prepared by addition of 200 µL of pure acetonitrile instead of the aflatoxin standard solutions plus 50 µL of bromoethanol. |
| ‡ Due to the toxicity of aflatoxins, special caution was taken in the preparation and handling of all reagents, nitrile gloves were used, the glassware was soaked in hypochlorite for deactivation of aflatoxins. All solvents and solutions were disposed of as hazardous waste. Work practices, safety equipment and facilities were designed to minimize the exposure of the operators and the environment to infectious microorganisms. A biosafety level 2 was set up when working with the mould cultures. |
| § Malt extract agar (Blakeslee) containing 0.6% of CAVASOL® (W7 1,8 Methyl-β-cyclodextrin, from Wacker, Münich, Germany) was sterilized and then 0.6% sodium deoxycholate from Sigma (USA) was added aseptically before plating on disposable Petri dishes (3.5 cm diameter, Deltalab, Spain). |
| ¶ Petri dishes containing modified malt extract agar (MACD) were inoculated with representative aflatoxigenic and non-aflatoxigenic strains and incubated at 32 °C. Excitation of the aflatoxigenic strains and acquisition of the RTP emission was achieved by means of a bifurcated optical fibre introduced inside the “dark box”, where the colonies cultured on the agar plates were placed. |
| || All luminescence measurements were carried our using a Cary Eclipse (Varian Iberica, Madrid, Spain) luminescence spectrometer equipped with a flash Xe arc source. Instrumental conditions for the acquisition of fluorescence spectra were: ex. wavelength 365 nm, ex. slit 10 nm, em. slit 10 nm, photomultiplier voltage 500 V. Instrumental conditions for the acquisition of the RTP spectra were: total decay time 0.020 s, excitation and emission slits were both fixed at 20 nm and the photomultiplier voltage was set at 600 V. The delay time (0.1 ms) and the gate time (1.00 ms) were optimized in order to avoid short-lived interfering emissions without collecting a high background. For in vivo measurements, RTP parameters were optimized to detect RTP emission from the aflatoxigenic strains in the minimum possible time. The photomultiplier voltage was set at 800 V and delay time was set at 0.1 ms. Longer delay times gave rise to lower background but also to a reduced sensitivity in the analysis of the aflatoxigenic strains (longer times required for positive analysis). |
| This journal is © The Royal Society of Chemistry 2006 |