Determination of the origin of urinary norandrosterone traces by gas chromatography combustion isotope ratio mass spectrometry
Moritz Hebestreit*a, Ulrich Flenkera, Gregor Fußhöllera, Hans Geyera, Ute Güntnera, Ute Marecka, Thomas Pipera, Mario Thevisa, Christiane Ayotteb and Wilhelm Schänzera
aInstitute of Biochemistry, German Sport University Cologne, Carl-Diem Weg 6, Cologne, Germany. E-mail: m.hebestreit@biochem.dshs-koeln.de; Fax: +49 221 4973236; Tel: +49 221 4982 5060
bMontréal Anti-doping Laboratory, Institut Armand-Frappier-Santé, 245, Boulevard Hymus, Pointe-Gaire, Québec H9R 1G6, Canada
First published on 28th July 2006
Abstract
On the one hand, 19-norandrosterone (NA) is the most abundant metabolite of the synthetic anabolic steroid 19-nortestosterone and related prohormones. On the other hand, small amounts are biosynthesized by pregnant women and further evidence exists for physiological origin of this compound. The World Anti-Doping Agency (WADA) formerly introduced threshold concentrations of 2 or 5 ng of NA per ml of urine to discriminate 19-nortestosterone abuse from biosynthetic origin. Recent findings showed however, that formation of NA resulting in concentrations in the range of the threshold levels might be due to demethylation of androsterone in urine, and the WADA 2006 Prohibited List has defined NA as endogenous steroid. To elucidate the endogenous or exogenous origin of NA, 13C/12C-analysis is the method of choice since synthetic 19-nortestosterone is derived from C3-plants by partial synthesis and shows δ13CVPDB-values of around −28‰. Endogenous steroids are less depleted in 13C due to a dietary mixture of C3- and C4-plants. An extensive cleanup based on two high performance liquid chromatography cleanup steps was applied to quality control and doping control samples, which contained NA in concentrations down to 2 ng per ml of urine. 13C/12C-ratios of NA, androsterone and etiocholanolone were measured by gas chromatography/combustion/isotope ratio mass spectrometry. By comparing δ13CVPDB-values of androsterone as endogenous reference compound with NA, the origin of NA in doping control samples was determined as either endogenous or exogenous.
Introduction
Anabolic steroids have been prohibited for use in sport by the International Olympic Committee (IOC) since the Olympic Games in Montréal in 1976. Nandrolone has been known as an anabolic agent since the 1930s1 and its use by athletes became popular in the late 1950s.2 It is mainly metabolized to conjugates of 19-norandrosterone (NA) and 19-noretiocholanolone (NE).3,4 Prohormones of nandrolone such as norandrostenediol or norandrostenedione likewise can be abused by athletes, but basically exhibit the same metabolism. The application of norethisterone which is present in contraceptives also leads to formation of NA. In these cases the major metabolite tetrahydronorethisterone can be found in contrast to illicit intake of anabolic 19-norsteroids.4 The question, whether urinary 19-norsteroids such as NA might be of endogenous origin has been discussed extensively. A review of this issue was published by Bricout and Wright.5 Recent findings showed that, besides the possible sources discussed (exercise, contaminated nutritional supplements, intermediates during the biological synthesis of estrogens etc.), a demethylating activity in urine can transform endogenous steroids such as androsterone (AND) or etiocholanolone (ETIO) into the corresponding 19-norsteroids (NA or NE).6 Amounts up to 2.2 ng of NA per ml of urine in doping control samples were due to this demethylating activity.7 Accordingly, the World Anti-Doping Agency (WADA) has revised the threshold concentration of 2 ng of NA per ml of urine4,8 and defined it an endogenous steroid.9 Thus, it is a pressing task to determine the origin of NA in urine in order to decide if its presence is due to the administration of nandrolone or a related substance or if it is of physiological origin due to e.g. the transformation of AND into NA. To differentiate between endogenous and synthesized naturally occurring steroids, the method of choice is gas chromatography combustion isotope ratio mass spectrometry (GC/C/IRMS), which was introduced to doping control in 1994.10 Synthesized steroids are normally made from Dioscorea spp. or soy.11 These are C3-plants, which are depleted in 13C in contrast to C4-plants. In consequence, synthesized steroids and their metabolites are also depleted in 13C in contrast to endogenous steroids since endogenous steroids derive from the diet, which is usually a mixture of C3-plants and C4-plants. Carbon isotope ratios are expressed as δ13CVPDB-values according to eqn 1 where R represents the 13C/12C molar ratio of the sample and of the VPDB-standard respectively. | (1) |
The challenge is to obtain δ13CVPDB-values from NA in urine at very low concentrations. δ13CVPDB-values down to 2 ng ml−1 should be confirmed whereas the lower limit for GC/C/IRMS-systems is about 10 ng of NA per injection to obtain valid 13C/12C-ratios. Thus at least 10 ml of urine have to be cleaned up efficiently, and the purity of the isolated target analytes is of utmost importance to guarantee baseline separation on the GC for reliable δ13CVPDB-values.12 Hence, a cleanup-method for 10 ml of urine was developed, which allows reliable 13C/12C-ratios of urinary NA down to 3 ng ml−1 of urine or even lower to be obtained. The δ13CVPDB-values can be compared with those from ETIO and AND which are isolated at the same time.
Experimental
Chemicals and standards
Methanol (puriss., distilled before use), acetone (GC grade) and methyl-tert-butylether (distilled before use) (MTBE) were purchased from KMF Laborchemie Handels GmbH (St. Augustin, Germany), n-hexane (gradient grade for liquid chromatography), isopropanol (p.a.) (IPA) and sodium phosphate (p.a.) from Merck (Darmstadt, Germany), β-Glucuronidase from E. coli from Roche Diagnostics (Mannheim, Germany) and acetonitrile (gradient grade for liquid chromatography) (AcN) from JT Baker (Deventer, Holland). The steroids AND and ETIO were purchased from Sigma-Aldrich (Steinheim, Germany) and the reference standard 5α-androstane-3α,17β-diacetate from Steraloids (Newport, USA). NA and its glucuronide were synthezised in our laboratory.13–15Method
The method comprises the following steps:1. Reversed phase solid phase extraction (RP SPE)
2. Enzymatic hydrolysis of glucuronides
3. Liquid–liquid extraction (LLE)
4. 1st normal phase high performance liquid chromatography (NP-HPLC) purification on a dimethylaminopropyl column: separation in two fractions
5. 2nd RP-HPLC purification on a C18 column: separation of one fraction in two subfractions
6. GC/C/IRMS
7. Interpretation of results
 | ||
| Fig. 1 NP-HPLC/UV chromatogram at 200 nm of 5 µg of NA, AND and ETIO each; collection pattern. | ||
 | ||
| Fig. 2 RP-HPLC/UV chromatogram at 200 nm of 5 µg of NA and AND each; collection pattern. | ||
Results and discussion
Validity of the method
 | ||
| Fig. 3 Relevant section of a GC/C-chromatogram of the NA-fraction of a blank urine; intensity m/z 44 on the left and the corresponding ratio 45/44 on the right panel. | ||
 | ||
| Fig. 4 Relevant section of a GC/C-chromatogram of the NA-fraction of a urine sample spiked with 4 ng of NA ml−1; intensity m/z 44 on the left and the corresponding ratio 45/44 on the right panel. | ||
| Concentration of NA/ng ml−1 | Mean CVPDB of NA ± standard deviation (‰) |
|---|---|
| 2 | −26.5 ± 0.35 (n = 5) |
| 4 | −26.8 ± 0.77 (n = 6) |
| 6 | −26.6 ± 0.73 (n = 9) |
| 8 | −26.8 ± 0.39 (n = 8) |
| Total | −26.7 ± 0.59 (n = 28) |
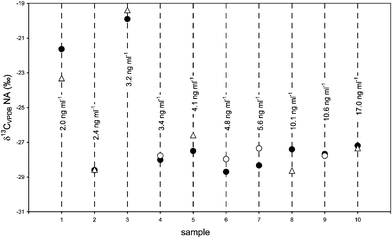 | ||
| Fig. 5 Repeated analyses of δ13CVPDB-values of NA in doping control samples and its concentration. First analysis under splitless conditions on the Optima δ3 column (●); second analysis COC on the Optima δ3 column (○) or on the HP-5MS column (Δ). | ||
 | ||
| Fig. 6 δ13CVPDB-values of NA vs. AND of control samples; the ellipses indicate endogenous or exogenous origin of NA. | ||
Fig. 7 shows the differences in the δ-values (expressed as Δδ) of AND and NA vs. the urinary concentration of NA of the control samples. Endogenous NA-concentrations caused by pregnancy did not exceed 2 ng ml−1 and the Δδ-values were lower than 2 ‰. NA-concentrations caused by oral administration of norandrostenediol or norandrostenedione of the selected urine samples were between 4 and 8 ng ml−1 and the Δδ-values did not fall below 4 ‰. By means of the Δδ-values, the origin of NA from control samples was classified accurately.
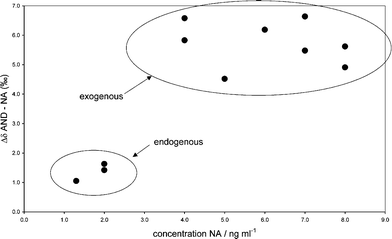 | ||
| Fig. 7 Δδ of AND and NA vs. the concentration of NA in control samples; the ellipses indicate endogenous or exogenous origin of NA. | ||
 | ||
| Fig. 8 δ13CVPDB-values of NA vs. AND of 25 doping control samples; 12 samples reanalysed under varying conditions; the ellipses indicate the presumable classification into endogenous or exogenous origin of NA. | ||
Fig. 9 shows the Δδ-values of AND and NA of the doping control samples vs. the concentration of NA, corrected for specific gravity if necessary.4 Δδ-values of the 12 samples showing exogenous δ13CVPDB-values for NA were larger than 4 ‰ and up to almost 9 ‰. They were identified over the whole concentration range. The differences in the δ13CVPDB-values between NA of endogenous origin and AND were below the WADA criterion of 3 ‰20 and did not exceed 2 ‰. The highest concentration for NA of endogenous origin was 5.6 ng ml−1. A steroids demethylating activity according to Grosse et al.6 was not detected in all of the samples with inconspicuous isotope signature. It was not detected in any of the samples showing exogenous origin of NA. No correlation depending on the gender of the athletes was observed. The dashed line in Fig. 9 indicates a possible decision limit to discriminate between endogenous and exogenous origin of NA. Hence, a doping offence with nandrolone or a corresponding 19-norsteroid was detected if the Δδ-value between NA and AND was above this limit.
 | ||
| Fig. 9 Δδ of AND and NA vs. the concentration of NA of 25 doping control samples (12 of them reanalysed under different conditions); Δδ-values above the possible decision limit indicated by the dashed line were deemed to show exogenous origin for NA whereas Δδ-values below this line were deemed to show endogenous origin for NA. | ||
Conclusions
The presented method, based on two HPLC cleanup steps, enables reliable measurements of the carbon isotope ratio of urinary NA traces by GC/C/IRMS under varying conditions. It was developed to elucidate the origin of urinary NA as either endogenous or exogenous to prove an abuse with nandrolone or corresponding 19-norsteroids. By means of the differences in the δ13CVPDB-values of NA and AND, the origin of NA was elucidated correctly as either endogenous or exogenous in control samples containing low amounts of NA of known origin. Accordingly, doping control samples were analysed and the origin of NA was elucidated as either endogenous or exogenous. Data of doping control samples are presented with an NA-concentration of up to 5.6 ng per ml of urine, but the carbon isotope ratios of NA indicate endogenous origin.Acknowledgements
This project was carried out with support of the WADA. Gratefully acknowledged are also the contributions of Vassilios Gougoulidis for the sample preparation and Yvonne Schrader for providing NA-containing urine samples of excretion studies.References
- A. Butenandt and K. Tscherning, Z. Physiol. Chem., 1934, 229, 167–182 Search PubMed.
- H. A. Haupt and G. D. Rovere, Am. J. Sports Med., 1984, 12, 469–484 Search PubMed.
- W. Schänzer, A. Breidbach, H. Geyer, C. von Kuk, E. Nolteernsting and M. Thevis, in Recent Advances in Doping Analysis. Proceedings of the 18th Cologne Workshop on Dope Analysis, 20th to 25th February 2000, ed. W. Schänzer, H. Geyer, A. Gotzmann and U. Mareck-Engelke, Sport und Buch Strauß, Köln, 2001, vol. 8, pp. 155–174 Search PubMed.
- WADA, Reporting Norandrosterone Findings. Technical Document TD2004NA, World Anti-Doping Agency, Montréal, Canada, 2004, http://www.wada-ama.org/rtecontent/document/nandrolone_aug_04.pdf Search PubMed.
- V. Bricout and F. Wright, Eur. J. Appl. Physiol., 2004, 92, 1–12 CrossRef CAS.
- J. Grosse, P. Anielski, P. Hemmersbach, H. Lund, R. K. Mueller, C. Rautenberg and D. Thieme, Steroids, 2005, 70, 499–506 CrossRef CAS.
- D. Thieme, P. Anielski, J. Grosse, P. Hemmersbach, H. Lund and C. Rautenberg, in Recent Advances in Doping Analysis. Proceedings of the Manfred Donike Workshop. 22nd Cologne Workshop on Dope Analysis, 7th to 12th March 2004, ed. U. Mareck, Sport und Buch Strauß, Köln, 2005, vol. 12, pp. 177–188 Search PubMed.
- WADA, The 2005 Prohibited List, World Anti-Doping Agency, Montréal, Canada, 2005, http://www.wada-ama.org/rtecontent/document/list_2005.pdf Search PubMed.
- WADA, The 2006 Prohibited List, World Anti-Doping Agency, Montréal, Canada, 2006, http://www.wada-ama.org/rtecontent/document/2006_LIST.pdf Search PubMed.
- M. Becchi, R. Aguilera, Y. Farizon, M.-M. Flament, H. Casabianca and P. James, Rapid Commun. Mass Spectrom., 1994, 8, 304–308 CAS.
- A. Kleemann and H. J. Roth, Arzneistoffgewinnung: Naturstoffe und Derivate, Thieme, Stuttgart, 1983 Search PubMed.
- A. Newman, Anal. Chem., 1996, 68, 373A–377A CAS.
- M. Thevis, G. Opfermann, H. Schmickler and W. Schanzer, J. Mass Spectrom., 2001, 36, 159–168 CrossRef CAS.
- W. Schänzer and M. Donike, Anal. Chim. Acta, 1993, 275, 23–48 CrossRef.
- W. Schänzer and M. Donike, in Recent Advances in Doping Analysis. Proceedings of the 12th Cologne Workshop on Dope Analysis, 10th to 15th April 1994, ed. M. Donike, H. Geyer, A. Gotzmann and U. Mareck-Engelke, Sport und Buch Strauß, Köln, 1995, vol. 2, pp. 93–112 Search PubMed.
- A. Schimmelmann, http://php.indiana.edu/%E2%88%BCaschimme/hc.html, 2004.
- U. Mareck-Engelke, G. Schultze, H. Geyer and W. Schänzer, in Recent Advances in Doping Analysis. Proceedings of the 18th Cologne Workshop on Dope Analysis, 20th to 25th February 2000, ed. W. Schänzer, H. Geyer, A. Gotzmann and U. Mareck-Engelke, Sport und Buch Strauß, Köln, 2000, vol. 8, pp. 145–150 Search PubMed.
- Y. Schrader and W. Schänzer, in Recent Advances in Doping Analysis. Proceedings of the 22nd Cologne Workshop on Dope Analysis, 7th to 12th March 2004, ed. W. Schänzer, H. Geyer, A. Gotzmann and U. Mareck, Sport und Buch Strauß, Köln, 2004, vol. 12, pp. 109–119 Search PubMed.
- Y. Schrader and W. Schänzer, unpublished work.
- WADA, Reporting and Evaluation Guidance for Testosterone, Epitestosterone, T/E Ratio and other Endogenous Steroids. Technical Document TD2004EAAS, World Anti-Doping Agency, Montréal, Canada, 2004, http://www.wada-ama.org/rtecontent/document/end_steroids_aug_04.pdf Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |