An electronic DNA microarray technique for detection and differentiation of viable Campylobacter species
Hai
Zhang
,
Zhilong
Gong
,
Odell
Pui
,
Yanming
Liu
and
Xing-Fang
Li
*
Environmental Health Sciences, Department of Public Health Sciences, University of Alberta, Edmonton, Alberta, Canada T6G 2G3. E-mail: xingfang.li@ualberta.ca; Fax: 1-780-492-7800; Tel: 1-780-492-5094
First published on 5th June 2006
Abstract
An electronic oligonucleotide microarray technique was developed for detection and differentiation of the viable Campylobacter species, C. jejuni, C. coli, and C. lari. This development consisted of four major components: identification of single nucleotide polymorphisms (SNPs) within the hsp60 gene as species markers, design of fluorescently labelled SNP-based reporters, development of an electronic microarray detection, and application of the integrated technique to analysis of Campylobacter species in food samples. A unique capability of this technique is the specific detection of viable cells and not dead ones. This is achieved by using mRNA of the 60 kDa heat-shock protein as the viability marker. The identification of two unique SNPs closely located at positions 291 and 294 of the hsp60 gene enabled the differentiation of the three Campylobacter species. This technique was able to detect as few as two viable Campylobacter cells. The analysis of 19 blind Campylobacter samples showed 100% agreement with their identities obtained using pulsed-field gel electrophoresis. The analysis of six chicken samples revealed the presence of C. coli in one of the samples.
Introduction
The Campylobacters C. jejuni, C. coli, and C. lari are the most frequent causes of bacterial gastroenteritis in humans.1,2 It is estimated that in the United States alone, Campylobacter species cause more than 2.4 million cases of diarrhoea annually.3Campylobacter species were isolated twice as frequently as Salmonella species from human infection cases.3 Although campylobacteriosis incidents are usually sporadic, outbreaks have occurred in recent years, even in affluent countries.3,4 Inadequately treated municipal water and untreated surface water due to run off of animal waste and improperly preparation/cooking of poultry have contributed to outbreaks.4–7 Epidemiological evidence suggests that animals, particularly poultry, cattle, wild birds, pigs, and domestic pets, are the key reservoirs for the strains infecting humans.3–7Fast, specific, and sensitive methods for biomonitoring of microbial pathogens are necessary. Culture methods and immunoassays are widely used; however, they require prolonged incubation, selective enrichment to reduce the growth of background flora, and biochemical identification. These methods are usually time-consuming and laborious. More importantly, the culture methods may show false-negative results due to cells entering a viable but non-culturable (VBNC) state.8,9 A number of alternative molecular techniques have been developed for identification of Campylobacter species, including conventional polymerase chain reaction (PCR),9–11 real time PCR,12 multiplex PCR,13,14 multi-locus sequence typing,15,16 and reverse transcription (RT)-PCR combined with either restriction enzyme digestion or restriction fragment length polymorphism.17,18 However, simultaneous detection and differentiation of C. jejuni, C. coli, and C. lari is still an analytical challenge because they have similar phylogenetic and genetic characteristics.19,20 Monitoring of specific species of Campylobacter is important for disease screening and epidemiological studies on which appropriate intervention and prevention strategies are based.13,21
DNA microarray techniques have been rapidly developed for gene expression analysis.22–27 These DNA microarray techniques generally involve gene amplification followed by hybridization analysis. They are particularly useful for detection of many genes in the whole genome of an organism. The common microarray approach relies on passive hybridization. A sample is applied to the entire gene chip regardless of the analytical requirements. Consequently, this technique is not versatile for analysis of multiple pathogens on the same chip.
Microelectronic oligonucleotide arrays could overcome this problem. Unlike the common cDNA arrays that are based on passive hybridization to the entire chip, the microelectronic array technique can actively transport DNA to, and hybridize at, discreet locations on the microchip.
The microelectronic array technique allows multiple independent analyses on the same array surface by selectively and independently targeting different DNA samples to specific locations using microelectronic addressing;28–30 however, only a few studies31–34 reported electronic microarray detection of pathogens, and none demonstrated specific detection of viable pathogens.
Other DNA microarray techniques35–42 have recently been developed for detection of specific pathogens. These techniques are based on detection of genomic DNA. They cannot differentiate live cells from dead cells. The information of viability of pathogens detected is important for tracking outbreaks and for the study of infectivity because only the live cells are infectious. No reported microarray technique is able to specifically detect viable microbial cells, while at the same time, differentiate Campylobacter species.
The primary objective of this study was to develop an electronic DNA microarray technique that enables the detection and speciation of the viable Campylobacter species jejuni, coli, and lari. Messenger RNA encoding the 60 kDa heat shock protein (hsp60) gene that is universally present in all microorganisms is only stable in viable cells and degrades rapidly when cells die.43 Thus, we chose hsp60 mRNA as the viability marker. We further identified single nucleotide polymorphisms (SNPs) with a highly conserved sequence of this mRNA in different Campylobacter isolates and determined SNPs that are specific to each of the target Campylobacter species. The analysis of these SNPs using the electronic microarray technique achieved the objective of differentiating closely related Campylobacter species.
Experimental
Materials
Chemicals (NaH2PO4, Na2HPO4, NaCl) that were used for preparation of buffer solutions were obtained from Fisher Scientific (Nepean, ON, Canada). The high salt buffer (pH 7.4) contained 50 mM NaH2PO4/Na2HPO4 with 500 mM NaCl and the low salt buffer contained 50 mM NaH2PO4/Na2HPO4 (pH 7.0). Histidine was obtained from Aldrich (Milwaukee, WI, USA). All the high and low salt buffers and L-histidine (50 and 100 mM) solutions were prepared in distilled de-ionized water (ddH2O) and filtered using 0.2 µm filters. The buffers were stored at 4 °C and the 100 mM histidine solutions were kept at −20 °C. The 50 mM histidine solutions were prepared once a week and kept at 4 °C. All chemical reagents used were of analytical grade. The 96-well plates used as sample vials in the microarray analysis were purchased from Nalgene Inc. (Rochester, NY, USA).Short DNA labelled with fluorescent Cy3 and Cy5 (reporters) and PCR primers were custom synthesized by Integrated DNA technologies (Coralville, IA, USA). Primers were desalted while reporters were purified using HPLC. They were all prepared in distilled deionized water (ddH2O) and stored at −20 °C.
Reagents and kits for RNA extraction, cloning, PCR, and RT-PCR, Trizol, Zero Blunt® PCR cloning kit, One Shot®E. coli TOP10 competent cells, Platinum® Taq DNA polymerase, SuperscriptII Reverse Transcriptase, and DNase I, were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). PCR purification kits (QIAquick PCR purification kit, QIAprep Spin Miniprep kit, and QIAquick Gel Extraction kit) were purchased from Qiagen (Valencia, CA, USA).
Campylobacter isolates were provided by the Agri-Food Systems branch of the Food Safety Division, Alberta Agriculture (Edmonton, Alberta, Canada). Other microbes used in the tests included E. coli O157∶H7, Vibrio cholerae O1, and Salmonella Typhi (provided by Dr Glen Armstrong at the Enterobacteria Culture Collection, Department of Medical Microbiology and Immunology), Cryptosporidium parvum (provided from Dr Mike Belosevic at the Department of Biological Sciences, University of Alberta), and non-pathogenic E. coli (ATCC 25922), Listeria monocytogenes, and Yersinia enterocolitica (provided by the Provincial Laboratory of Public Health (Microbiology) at the University of Alberta).
Biosafety
Experiments involving pathogenic microbes were performed according to the standard procedures for biohazards. Prior to the study, the personnel involved in this study were trained and certified by the Biosafety Office at the University of Alberta. The handling and preparation of all cultures and samples was carried out strictly in biohazard hoods. The wastes from the experiments were autoclaved and disposed of according to the biosafety procedures.Campylobacter cultures and RNA extraction
All Campylobacter isolates used in this study were provided by the Agri-Food Systems Branch of the Food Safety Division, Alberta Agriculture, where these isolates were previously analyzed using pulsed-field gel electrophoresis (PFGE). These isolates were inoculated in sterile tryptic soy broth to a 3.0 McFarland density and they grew to approximately 9 × 108 colony forming units (CFUs) mL−1. An aliquot of 15 µL of each culture in tryptic soy broth was transferred to a 1.5 mL centrifuge tube, and incubated at 48 °C in a water bath for 25 minutes to heat shock the organisms and induce hsp60 mRNA. The cell pellets were collected after 10 min of centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g at 4 °C. The supernatant was removed.
000 g at 4 °C. The supernatant was removed.
The total RNA in the cell pellets was extracted using Trizol reagent (Invitrogen) following the recommended procedures. The concentration and purity of the extracted RNA was measured using a SmartSpecTM 3000 spectrophotometer (BioRad, Hercules, CA, USA). The RNA extracts we obtained had a ratio of absorbance (A260/A280) greater than 1.8, indicating minimum DNA and proteins in these samples. These RNA extracts were immediately frozen at −80 °C until use.
Twelve Campylobacter isolates, including three C. coli, three C. lari, and six C. jejuni, were initially used to obtain the sequence of the mRNA of the hsp60 gene for identification of the unique sequence fragment and single nucleotide polymorphisms (SNPs). The other two groups of isolates were analyzed to evaluate the RT-PCR/electronic microarray technique described below.
RT-PCR
Fragments of the mRNA encoding the hsp60 gene, 460 bp (from 54 to 513) of C. jejuni and C. coli and a fragment of 446 bp (from 54 to 499) of C. lari, were amplified. The RNA extracts from the twelve Campylobacter isolates were reverse-transcribed and PCR amplified using the designed primers shown in Table 1. Prior to the RT reactions, the total RNA extracts from cells, including three of C. coli, three of C. lari, and six of C. jejuni, were treated with DNase I to eliminate DNA contamination. Duplicates of RNA samples and controls were prepared, and RT reactions were performed using Superscript Reverse Transcriptase. The RT reaction mixtures consisted of 11 µL of the DNase-treated RNA solution or ddH2O as negative control, 0.6 µL of gene-specific reverse primer mix (containing all three species-specific reverse primers, 5 µM of each; and their sequences are shown in Table 1), 0.4 µL of 25 mM dNTP, 4 µL of 5× first-strand buffer, 2 µL of 0.1 M DTT, and 1 µL of RNaseOUT recombinant ribonuclease inhibitor (40 units µL−1). The mixed solution was incubated at 42 °C for 2 min. To the reaction mixtures, an aliquot of 1 µL (200 units) of Superscript Reverse Transcriptase was added to the treated RNA samples. To the control, an aliquot of 1 µL of ddH2O was added. The reaction mixtures and DNase controls were incubated at 42 °C for 50 min, and heated at 70 °C for 15 min. The cDNA was used as a template for PCR amplification.| Primer | Sequence | T m/°C | |
|---|---|---|---|
| C. jejuni | Forward | 5′-GCA GGT GCA AAT CCT ATC G-3′ | 63.2 |
| Reverse | 5′-AAG TTG CAA GCG CTT CAC C-3′ | 65.4 | |
| C. coli | Forward | 5′-GCT GGA GCA AAT CCT ATC G-3′ | 62.3 |
| Reverse | 5′-AAG TTG CAA GTG CTT CAC C-3′ | 60.5 | |
| C. lari | Forward | 5′-GCA GGT GCT AAT CCT ATC G-3′ | 59.8 |
| Reverse | 5′-TCA CCT TCA ATG TCT TCA GC-3′ | 60.4 |
The targets of 460 bp of C. jejuni and C. coli, and 446 bp of C. lari were amplified using Platinum® Pfx DNA polymerase. The RT solution described above was used directly as the template, and an equal volume of ddH2O was used as the template for PCR negative control. Each polymerase chain reaction contained the following: 5 µL of 10× Pfx Amp buffer, 1 µL of 50 mM MgSO4, 6 µL of 2.5 mM dNTP mixture, 1.5 µL of 10 µM of the forward primer, 1.5 µL of 10 µM of the reverse primer, 1.0 µL of Pfx DNA polymerase (2.5 units), 2 µL of the template, and 32 µL of sterile ddH2O for a total volume of 50 µL. The reaction mixture was incubated at 94 °C for 3 min, followed by 40 cycles of PCR amplification using the following temperature program: denaturation at 94 °C for 15 s, followed by annealing for 30 s at 58.2 °C for C. jejuni, 50.5 °C for C. coli, and 54.8 °C for C. lari, and extension at 70 °C for 30 s with a final extension step at 72 °C for 10 min. The amplification was performed in a PTC-100Tm Programmable thermal controller (MJ Research, Waltham, MA, USA).
Cloning and sequencing of the hsp60 gene from different isolates
Prior to cloning, the PCR products were separated using agarose gel electrophoresis at 0.5 V cm−1 overnight. The desired DNA fragments were excised from the gel, extracted, and purified using a QIAquick Gel Extraction Kit following the manufacturer recommended protocol. The purified PCR products were cloned using a Zero Blunt® PCR cloning kit, then sequenced using a Beckman Coulter CEQ2000XL DNA sequencing system (Beckman Coulter, Fullerton, CA, USA) with M13 universal primers in the Department of Biochemistry, University of Alberta. The Sanger dideoxynucleotide termination sequencing protocol was used. Thermo-cycle sequencing reactions were resolved and detected through a single lane of electrophoresis. Duplicate plasmid samples of the Campylobacter isolates (three of C. coli, three of C. lari, and six of C. jejuni) were sequenced, and the parallel sequencing results were obtained and compared with those in the SWISS-PROT database. The sequence results were used to develop a microarray detection method.Preparation of the targets and design of the reporters for microarray detection
The Nanogen microarray detection uses two fluorophores, Cy3 (λex 550 nm and λem 567 nm) and Cy5 (λex 635 nm and λem 667 nm). In order to use two colors to simultaneously differentiate three Campylobacter species, two PCRs based on the target selections were designed, as shown in Fig. 1: PCR 1 using Set-1 primers amplifies the 5′ to 3′ strand of C. lari, and PCR 2 using Set-2 primers amplifies the 3′ to 5′ strands of C. jejuni and C. coli. Two sets of primers (Table 2) were designed and used for PCR amplification reactions 1 and 2. | ||
| Fig. 1 Schematic diagram showing two PCRs to amplify the targets for microarray detection. Complementary DNA reverse transcribed from hsp60 mRNA is the template. In PCR 1 using primers set-1, the strand from 5′ to 3′ is amplified and labelled with biotin, which is for detection of C. lari. The PCR 2 using the primers set-2 the strands of 3′ to 5′ are amplified and labelled with biotin. The PCR products of C. coli and C. jejuni are reported by the corresponding reporters-coli and jejuni. Solid circle refers to biotin-label. | ||
| Primer | Sequence | T m/°C | |
|---|---|---|---|
| a F: forward primer; R: reverse primer | |||
| Set-1 | F | 5′-/Bio/-GAA GGT ATG CAA TTT GAC AG-3′ | 50.3 |
| R | 5′-TCA CCT TCA ATA TCT TCA GC-3′ | 50.7 | |
| Set-2 | F | 5′-GAA GGT ATG CAA TTT GAC AG-3′ | 50.3 |
| R | 5′-/Bio/-TCA CCT TCA ATA TCT TCA GC-3′ | 50.7 | |
PCR amplification using Taq polymerase was performed under the following conditions: initial denaturation at 94 °C for 3 min followed by 40 cycles of denaturation at 94 °C for 15 s, annealing at 45 °C for 30 s and extension at 72 °C for 30 s, and final extension at 72 °C for 10 min. An aliquot of 2 µl of the cDNA produced above was used as the template for the individual polymerase chain reactions. During the method development, PCR products from each PCR were separated and visualized on 1.5% agarose gel, and desalted by using a QIAquick PCR purification kit prior to microarray analysis. The final concentration of the target was 1.4 nM for DNA microarray analysis. The gel separation step was not required after the technique was developed.
For microarray detection of the targets, the reporters were specifically designed according to the sequences of the targets. The reporters of C. lari and C. jejuni were labelled with Cy3, while the reporter of C. coli was labelled with Cy5. Although both C. lari and C. jejuni reporters were labelled with the same Cy 3 dye, C. lari and C. jejuni could be differentiated because their reporters recognized different sequences of the PCR products from reaction 1 (for C. lari) and 2 (for C. jejuni) as described above. The sequences and melting temperatures of the reporters are summarized in Table 3. The nucleotides shown in bold in the reporters correspond to the SNPs in the targets that were identified as species markers.
| Reporter | Sequence | T m/°C |
|---|---|---|
| R-jejuni | 5′-Cy3-TAT CAC TAA TGC A-3′ | 32.6 |
| R-coli | 5′-Cy5-ATA ACC AAT GCA-3′ | 32.4 |
| R-lari | 5′-Cy3-ATT TGT AAT GAA ATA T-3′ | 32.1 |
Microarray instrumentation
A NanoChip™ workstation (Nanogen, San Diego, CA, USA) was used as a platform to facilitate this study. The NanoChip™ workstation consists of a Nanochip™ cartridge, a loader, and a reader. A semiconductor microchip is located within the cartridge. It consists of 100 individual test sites, each wired to an individual metal contact at the back of the chip. The semiconductor is coated with streptavidin incorporated in a hydrogel permeation layer. The loader was used to electronically deliver the negatively charged DNA molecules from a 96-well plate to the designated sites on a chip according to a user-defined map. The loader was also used to wash the chip with 50 mM L-histidine solution. The reader was used to detect the fluorescence signals from the reporters that were hybridized with complementary and biotinylated target DNA bound to streptavidin on the chip. The reader also facilitated the use of elevated temperature and low salt washing to minimize cross hybridization and background.The electronic microarray detection process is described briefly here. Heat denatured biotinylated target DNA molecules obtained through RT-PCR were prepared in 50 mM L-histidine, and were electronically delivered onto a microarray chip by the loader. The biotinylated strands were bound onto the testing sites through the biotin–streptavidin interaction. Unbound DNA was washed off by 50 mM L-histidine using the loader. After incubation with species-specific fluorescently labelled reporters in high salt buffer (50 mM NaH2PO4–Na2HPO4, pH 7.4 with 500 mM NaCl), the fluorescence signals were scanned and recorded using the reader at the optimized conditions. The optimized parameters were 2.0 V for 140 s for target loading on the loader, and 29 °C for thermal stringency on the reader.
The targets were identified based on three pieces of information: the specific reporters recognizing the products from the two sets of the PCRs, the color of the reporters, and the intensity of the fluorescence signal. The data acquisition and analysis were obtained using the built-in software. The threshold for a positive identification was a signal-to-background ratio higher than 3.
Preparation of chicken rinse samples
Six retail air-chilled whole broil chickens were randomly purchased from three different grocery stores in Edmonton, Alberta, Canada, and kept on ice for delivery to our laboratory. Collection of chicken rinse samples for bacterial analysis was conducted according to the USDA/FSIS Microbiology Laboratory Guidebook.44 Briefly, a fresh chicken was placed in a large sterile plastic bag (Stomacher™ 3500 bag), and 200 mL of 0.1% peptone water was added. The bag was twisted to seal it and the contents were shaken for 2 minutes. A corner of the bag was cut, and the rinse solution was drained and filtered through sterile double-layered cheesecloth and collected in a sterile 250 mL container. The procedures were performed in an aseptic manner. The collected rinse solution was incubated in a 48 °C water bath for 25 min to heat shock the microbial cells, and then centrifuged at 4 °C using 16000 g for 15 min. The supernatant was discarded. The pellet was collected and suspended in 5 mL of Trizol reagent and homogenized by vortexing. A 1 mL aliquot of the homogenized sample was used for the extraction of total RNA following the procedures described above. Desalted PCR products from each reaction were completely used for the analysis on the microarray.
Results
Identification of species-specific SNPs
In order to develop a SNP-based technique that can differentiate C. jejuni, C. coli, and C. lari, the SNPs in the hsp60 gene of these three species were first identified. The identification of SNPs was carried out by comparing specific sequences of these three microbes in the GenBank. The sequence information in the Genbank was further supplemented by sequencing the cDNA of a 460 nucleotide (nt) region of hsp60 mRNA from 3 isolates of C. lari, 3 isolates of C. coli, and 6 isolates of C. jejuni. The sequencing results from the 12 isolates were aligned and compared with those in the GenBank. A unique region (274–300 nt) of the sequences from the representative isolates is shown in Scheme 1. Differences in the sequences within this region of the hsp60 gene in C. jejuni, C. coli, and C. lari were detected at positions 282, 288, 291, 294, 298, and 300. Interestingly, two SNPs located at positions 291 and 294 are unique to the individual species. At position 291, C is unique to C. jejuni, A is unique to C. coli, and T is unique to C. lari. At position 294, T is unique to C. jejuni, C is unique to C. coli, and A is unique to C. lari. From the alignment of all the sequences of the hsp60 gene from all the 12 isolates with those in the GenBank, we found that the base compositions at positions 291 and 294 were identical within the same species but distinctly different between different species. Therefore, SNPs at 291 and 294 were chosen as characteristic markers to differentiate the three Campylobacter species.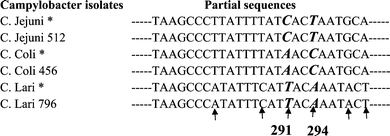 | ||
| Scheme 1 Alignment of partial sequence of the hsp60 gene identifying the SNPs unique to the species of Campylobacter. Note: Species specific SNPs are shown in bold and italic. * The sequences from GenBank. ↑ Differences in sequences between Campylobacter species were found. | ||
Specificity of reporters
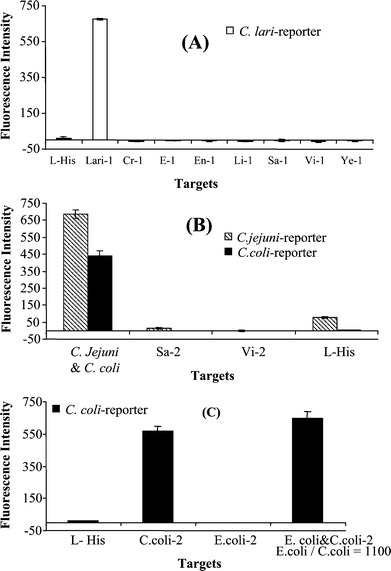
To detect the three Campylobacter species, fluorescently labelled oligonucleotide reporters (Table 3) were designed to recognize the specific targets carrying the SNPs located at 291 and 294. The biotinylated cDNA targets of C. coli, C. jejuni, and C. lari that were produced by the PCRs 1 and 2 (Fig. 1) were electronically delivered to separate spots (triplicate spots for each target) on the chip. The reporter-lari was designed to specifically recognize the target of C. lari. When this reporter was applied to the spots that were addressed with individual targets of C. coli, C. jejuni, and C. lari, the expected green fluorescence from Cy-labelled reporter was detected only from the spots that were addressed with C. lari, and background was observed from in the other spots addressed with C. coli and C. jejuni, as shown in Fig. 2A. These results demonstrate that the reporter-lari specifically recognizes the target C. lari and not the closely related C. coli and C. jejuni. Similar experiments were performed to examine the specificity of the reporter-coli and reporter-jejuni, in which the mixture of the reporter-coli (red, Cy5) and reporter-jejuni (green, Cy3) were applied to the targets of C. coli, C. jejuni, and C. lari (Fig. 2B). As expected, green fluorescence was evident only from the spots addressed with C. jejuni, red fluorescence was detected from the spots addressed with C. coli, and neither red nor green fluorescence was detected from the target of C. lari. These results suggest that each of the reporters specifically hybridize to its corresponding target. Although the targets of C. jejuni and C. coli had only one nucleotide difference (Scheme 1), they were clearly differentiated using the SNP-based reporters for C. jejuni and C. coli, respectively. The spots addressed with L-histidine buffer were used to measure the background fluorescence. Fig. 2 clearly demonstrates that the three SNP-based reporters are specific to the individual species of Campylobacter. | ||
| Fig. 2 Testing specificity of (A) the C. lari reporter and (B) the C jejuni and C. coli reporters. Biotinylated PCR products of the three species (C. lari, C. jejuni, and C. coli) were separately addressed onto different spots on the chip (triplicate spots for each target). (A) Only C. lari reporter (green) was applied for hybridization (each target concentration: 4 nM). (B) Both C. jejuni and coli reporters (red for coli and green for jejuni) were used for hybridization (each target concentration: 27 nM). | ||
There was no interference from other microorganisms when the reporter sequences were searched against those in the GenBank. Several common food-borne and water-borne pathogens, including E. coli O157∶H7, non-pathogenic E. coli (ATCC 25955), Salmonella Typhi (R704101), Vibrio cholerae O1, Listeria monocytogenes (R1694101), Yersinia enterocolitica (K1332/03), and Cryptosporidium parvum, were used to further examine whether they interfered with the detection of the target Campylobacter species. The RNA was separately extracted from the microbial cells and was prepared using the RT-PCR amplification. The PCR 1 products of these microbes were first analyzed by gel electrophoresis (results not shown). This analysis could not differentiate Campylobecter from Vibrio cholerae O1 and Salmonella Typhi. However, the microarray detection of these products (shown in Fig. 3A) only detected the target C. lari, and no positive signals from any other test pathogens including Vibrio cholerae O1 and Salmonella Typhi, confirming that these microorganisms did not interfere with the microarray detection of the target Campylobacter species using the specific reporters. Similarly, Fig. 3B shows the microarray detection of PCR 2 products of S. Typhi, V. cholerae, C. jejuni, and C. coli using the C. jejuni-reporter and the C. coli-reporter. The results demonstrate that this technique specifically detects the correct targets C. jejuni and C. coli and that S. Typhi and V. cholera do not interfere with the detection of these targets.
 | ||
| Fig. 3 Evaluation of interference of different pathogens with the microarray detection of the target Campylobacter using specific reporters. (A) No interference with the detection of C. lari was detected from Cryptosporidium parvum (Cr-1), E.coli O157∶H7 (E-1), non-pathogenic E.coli (ATCC 25955) (En-1), Listeria monocytogenes (R1694101) (Li-1), Salmonella Typhi (R704101) (Sa-1), Vibrio cholerae O1(Vi-1), and Yersinia enterocolitica (K1332/03) (Ye-1) using the C. lari-reporter. The fluorescence signals were obtained from the PCR 1 products of these microorganisms. The target concentrations were C. lari 8.5 nM, Vibrio cholerae O1 7.7 nM, and the others were 10 nM. (B) No interference was detected from Salmonella and Vibrio using the C. jejuni-reporter and C. coli-reporter. The fluorescence signals were obtained from the PCR 2 products of Salmonella Typhi, Vibrio cholerae O1, C. jejuni, and C. coli. (Target concentrations: C. jejuni: 29.7 nM; C. coli: 40.9 nM; Salmonella Typhi and Vibrio cholerae O1: 6.2 nM each). (C) Microarray detection of C. coli, E. coli O157∶H7, and the mixture when E. coli O157∶H7 was present 1100 times higher than the target C. coli, showing no interference from E. coli O157∶H7 with the detection of the target. Triplicate spots were used for each sample. | ||
In a real sample, other common bacteria are often present at a much higher population than the target microorganism. To evaluate the ability of this technique to detect the target Campylobacter when other microorganisms are present at much higher levels, C. coli was used as the target and E. coli O157∶H7 as the non-target bacterium. In this test, a mixture of total RNA of E. coli O157∶H7 (54.4 ng µL−1) and C. coli 456 (0.049 ng µL−1) were analyzed by the RT-PCR microarray technique. The non-target E. coli O157∶H7 was present at over 1000-fold higher levels than the target C. coli.Fig. 2C shows the fluorescence signals obtained from a control, C. coli alone, E. coli O157∶H7 alone, and the mixed targets of C. coli and E. coli O157∶H7. The signals of C. coli and the mixture of C. coli and E. coli were 560 and 645 (S/B = 59), respectively, whereas the signals obtained from E. coli O157∶H7 alone were at the background level (S/B = 1). These results correctly identified the target, C. coli, in the pure and mixed bacterial samples without interference from the non-target, even when the level of the non-target present in the same sample was over 1000-fold higher than the target. This demonstrates the capability of this technique for specific detection of the target Campylobacter in a mixed population of microorganisms.
Specific detection of live but not dead cells
To demonstrate the unique capability of this technique that detects live cells but not dead cells, a live culture sample and a heat-killed culture sample were analyzed. To generate a dead cell sample, a culture of 0.5 mL of C. coli isolate 456 (9 × 108 CFU mL−1) was heated at 100 °C in a water bath for 15 min. The heat-killed cells were kept at room temperature for 2 days before the total RNA was extracted from these cells. This procedure was based on the experiment conducted by Sheridan et al.43Fig. 4 shows the microarray detection of killed C. coli cells (4.5 × 108) and positive controls of C. coli, C. lari, and C. jejuni. Only background signals were observed from the dead cell samples, demonstrating that the dead cells were not detected. The undetectable level of mRNA in the dead cells agrees with that reported by Sheridan et al.,43 in which the groEL mRNA (hsp60 mRNA) of E. coli became undetectable after 2 h incubation at room temperature following the heat killing. The live cells of C. coli, C. lari, and C. jejuni were correctly identified (Fig. 4). These results demonstrate that this RT-PCR microarray detection technique can specifically detect viable cells but not dead cells. The ability to detect viable cells but not dead ones is important because only viable cells pose a potential threat to public health.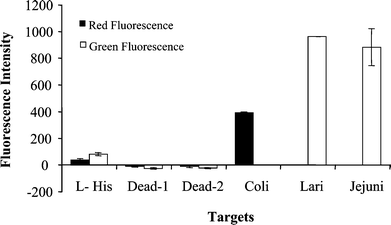 | ||
| Fig. 4 Detection of viable cells of C. coli, C. lari and C. Jejuni and dead cells of C. coli. Preparation of dead cells: 4.5 × 108 cells of C. coli in 0.5 mL in water were boiled for 15 min and kept at room temperature for 2 days before RNA extraction. All the other procedures involved in the analysis are the same as those described in the Experimental. L-His: L-histidine; Dead-1: the target of dead cell prepared using the PCR 1; Dead-2: the targets of dead cells prepared using the PCR 2; The targets of C. coli, C. lari and C. Jejuni were prepared from live cells as positive controls. Target concentrations for C. coli, C. lari, and C. jejuni were 50 nM. Triplicate spots were used for each sample. | ||
Sensitivity
Two sets of experiments were conducted to evaluate the sensitivity of the RT-PCR microarray detection technique. In the first set of experiments, various amounts (23 fg to 25 pg) of total RNA samples of C. jejuni were used as the starting materials. These RNA samples were prepared through serial dilution of 2046 ng RNA that was extracted from a C. jejuni culture (9 × 107 cells). The RNA samples were reverse transcribed to cDNA, and the targets were amplified and detected using the microarray technique. The samples containing 0.2 to 25 pg of total RNA (equivalent to 9–1100 cells) produced sufficient S/B ratio of greater than 8. Further dilution of the RNA samples to 23–246 fg (equivalent to 1–10 cells) also generated sufficient S/B (>4) for detection of the target. The total RNA at as low as 23 fg (equivalent to 1 cell) provided positive detection of C. jejuni, demonstrating the high sensitivity of this technique.The second set of experiments was performed with serially diluted cell samples containing 2 × 104, 2 × 103, 200, 100, 60, 30, 8, and 2 cells. The analysis of the PCR products of the samples containing more than 30 cells produced S/B ratios greater than 4, positively detecting the target. The threshold for a positive detection was S/B > 3. The analysis of triplicate samples supposedly containing 8 to 9 cells, one was positive and two were negative. The analysis of seven replicate samples supposedly containing 1 to 2 cells from serial dilution show the presence of the target cells in two samples and absence in the other five samples. The negative results in the five samples is not due the inability of the method for detecting 1 to 2 cells, rather they reflect the probability in obtaining 1–2 cells from serial dilutions. The presence or absence of a single cell in a given diluted sample may be governed by the Poisson distribution. The negative results are due to the absence of the target cells. Replicate negative controls consistently show the S/B value below 3, indicating the absence of the target cells. These results demonstrate the potential of this technique to detect a small number of cells. The minimum infection dose for Campylobacter is ∼500 cells. The sensitivity of the RT-PCR microarray technique is sufficient for the detection of Campylobacter below the infectious dose.
Intra-laboratory validation
To further validate this technique, seven samples were prepared with different combinations of RNA samples from the isolates C. jejuni-782, C. lari-803, and C. coli-456 in our laboratory. These samples were tested in a blind fashion for intra-laboratory validation. Each blind sample contained either one or more than one species with a total concentration of 107 CFU mL−1 in 1 mL. The identification of the isolate and mixed isolates in these blind samples was 100% in agreement with their identities.Detection of different Campylobacter isolates
To further demonstrate the specificity and applicability of the SNPs as species markers, 19 samples of unknown Campylobacter isolates were obtained from the Alberta Agri-Food Systems Branch of the Food Safety Division. Seven of these samples were analyzed by a graduate student who developed the technique and twelve samples were analyzed by another student who received training of the technique. The two separate tests were carried out using the RT-PCR microarray detection in a double-blind fashion. The total RNA from individual isolates was extracted and amplified using RT-PCR and the PCR products were analyzed using the SNP-based microarray detection described above. The results of these blind isolates are shown in Table 4. The identification of all the isolates completely matched those previously identified by PFGE conducted at the Alberta Agri-Food Systems Branch of the Food Safety Division, except for the isolate 678. The sequencing analysis of the PCR products of isolate 678 confirmed that it was C. jejuni, demonstrating that the SNP-based microarray detection is more specific than PFGE. These results confirm that the unique SNPs at 291 and 294 may be used as species markers for speciation of C. jejuni, C. coli, and C. lari. This is the first report identifying the unique SNPs at 291 and 294 in the hsp60 gene of these Campylobacter species and their applications as species-markers for microarray detection.| Isolate no | S/B | Results by this method | Sample identity by PFGE | Match | |
|---|---|---|---|---|---|
| a R = red, G = green, P1 = products of PCR 1, P2 = products of PCR 2. Threshold for positive call is S/B > 5. Y = yes, match. | |||||
| Analyst 1 | 61 | 5.4 (P2G) | C. jejuni | C. jejuni | Y |
| 98 | 20 (P2G) | C. jejuni | C. jejuni | Y | |
| 123 | 5.4 (P2G) | C. jejuni | C. jejuni | Y | |
| 143 | 18 (P2R) | C. coli | C. coli | Y | |
| 181 | 5.7 (P2G) | C. jejuni | C. jejuni | Y | |
| 11y | 92 (P2R) | C. coli | C. coli | Y | |
| 790 | 38 (P1G) | C. lari | C. lari | Y | |
| Analyst 2 | 432 | 7.3 (P2G) | C. jejuni | C. jejuni | Y |
| 456 | 21 (P2R) | C. coli | C. coli | Y | |
| 543 | 7.2 (P2G) | C. jejuni | C. jejuni | Y | |
| 577 | 7.6 (P2G) | C. jejuni | C. jejuni | Y | |
| 701 | 19 (P2R) | C. coli | C. coli | Y | |
| 773 | 25 (P1G) | C. lari | C. lari | Y | |
| 777 | 17 (P2R) | C. coli | C. coli | Y | |
| 779 | 26 (P1G) | C. lari | C. lari | Y | |
| 782 | 6.3 (P2G) | C. jejuni | C. jejuni | Y | |
| 790 | 28 (P1G) | C. lari | C. lari | Y | |
| 796 | 27 (P1G) | C. lari | C. lari | Y | |
| 678 | 8.6 (P2G) | C. jejuni | C. coli | N | |
Analysis of chicken samples
The application of this technique for screening microbial contamination in food was further demonstrated by analysis of chicken samples obtained from grocery stores. Previous epidemiological studies found that the prevalence of Campylobacter in Edmonton retail chicken was approximately 58%.45 Based on the published method for estimation of the required sample size (the number of samples required which will result in at least one positive result occurs),46 six samples were required in order to find a positive unit with a probability of 0.95. Therefore, six retail air-chilled whole broil chickens were randomly purchased from three grocery stores in Edmonton. No Campylobacter was detected in five of the six chicken samples. One chicken sample was detected to contain C. coli. This sample was repeatedly tested and C. coli was consistently detected. Table 5 presents the microarray results of these chicken samples.| Sample no. | Signal-to-background ratio | Identified Campylobacter | |||
|---|---|---|---|---|---|
| PCR 1a | PCR 2b | ||||
| Green | Red | Green | Red | ||
| a PCR 1 using green reporter for identification of C. lari. b PCR 2 using green reporter for identification of C. jejuni and red reporter for C. coli. | |||||
| 1 | −0.30 | −0.34 | −0.36 | −0.04 | ND |
| 2 | −0.39 | 0.00 | −0.49 | −0.11 | ND |
| 3 | −0.42 | −0.13 | −0.52 | −0.15 | ND |
| 4 | −0.40 | 0.72 | −0.52 | −0.13 | ND |
| 5 | 1.05 | 0.69 | 1.27 | 5.99 | C. coli |
| 6 | 1.04 | 0.41 | 1.97 | 0.75 | ND |
Discussion
Campylobacter jejuni and C. coli are the leading causes of bacterial gastroenteritis. The infection is zoonotic, with many animals, such as chickens, cattle, and pigs, acting as reservoirs for this organism. Routes of transmission to humans include ingestion of undercooked poultry or other meat, unpasteurized milk, and contaminated water. Current biochemical techniques are unreliable for speciation of Campylobacter species, which has implications for epidemiological tracing of the source of infection and antimicrobial regimens. Campylobacter species, especially those subject to harsh conditions in environmental samples, may enter a viable but non-culturable (VBNC) state, which can lead to false-negative culture results. The molecular techniques offer increased sensitivity and decreased turnaround time. Previous methods have employed DNA as a marker, but RNA more accurately reflects the viability of the organism because RNA degrades rapidly following cell death.These problems were addressed by integrating a reverse transcription polymerase chain reaction (RT-PCR) with an SNP-based microarray detection technique. Unique SNPs were identified as species markers. The technique uses the mRNA encoding the 60 kDa heat shock protein as the target for RT-PCR amplification, and combines with SNP-based microarray speciation of the Campylobacter species using the species-specific reporters we designed. This method can reliably discriminate between C. jejuni, C. coli, and C. lari. Two different analysts obtained accurate identification of the isolates, demonstrating the reproducibility and reliability of the technique. This technique is fast compared to the conventional methods involving culture steps. This technique can provide results within 8 hours from the RNA extraction to microarray analysis, whereas the conventional techniques involving culturing require days. The correct identification of 19 different isolates demonstrates the potential use of the two unique SNPs as biomarkers of C. jejuni, C. coli and C. lari. The integration of RT-PCR with microarray detection provides specificity, high sensitivity, and the unique capability of detecting viable cells and speciation of Campylobacter species. Furthermore, the method successfully detected the presence of bacteria in retail chicken samples without culturing steps. Other potential applications of this technique include analysis of bacterial contamination in food and water samples as well as clinical diagnosis of bacterial infection.
Acknowledgements
This study was financially supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canadian Water Network (CWN), Alberta Health and Wellness, an Alberta Heritage Foundation for Medical Research (AHFMR) Summer Research Studentship (to OP), and a NSERC University Faculty Award (to XFL). The authors would also like to thank Margaret Mcfall, Carol Goertz, and their colleagues at the Agri-Food Systems Branch of the Food Safety Division, Alberta Agriculture, Edmonton, Alberta, for generously providing the Campylobacter isolates.References
- J. P. Butzler, Y. Glupcynski and H. Goossens, Curr. Opin. Infect. Dis., 1992, 5, 80–87 Search PubMed.
- A. J. Lastovica and M. B. Skirrow, in Campylobacter, ed. I. Nachamkin and M. J. Blaser, American Society for Microbiology, Washington, DC, 2000; pp. 89–120 Search PubMed.
- C. R. Friedman, J. Neimann, H. C. Wegener and R. V. Tauxe, in Campylobacter, ed. I. Nachamkin and M. J. Blaser, American Society for Microbiology, Washington, DC, 2000, pp. 121–138 Search PubMed.
- S. E. Hrudey and E. J. Hrudey, Safe Drinking Water: Lessons from Recent Outbreaks in Affluent Nations, IWA Publishing, London, UK, 2004, pp. 81–423 Search PubMed.
- C. Thomas, H. Gibson, D. J. Hill and M. Mabey, J. Appl. Microbiol., 1999, 85, 168S–177S.
- R. Nannapaneni, R. Story, K. C. Wiggins and M. G. Johnson, Appl. Environ. Microbiol., 2005, 71, 4510–15 CrossRef CAS.
- P. Padungton and J. B. Kaneene, J. Vet. Med. Sci., 2003, 65, 161–70 Search PubMed.
- J. L. Tholozan, J. M. Cappelier, J. P. Tissier, G. Delattre and M. Federighi, Appl. Environ. Microbiol., 1999, 65, 1110–1116 CAS.
- D. M. Rollins and R. R. Colwell, Appl. Environ. Microbiol., 1986, 52, 531–538 CAS.
- T. A. Burnett, M. A. Hornitzky, P. Kuhnert and S. P. Djordjevic, FEMS Microbiol. Lett., 2002, 216, 201–209 CAS.
- L. Dedieu, J.-M. Pagès and J.-M. Bolla, J. Clin. Microbiol., 2004, 42, 2301–2305 CrossRef CAS.
- S. T. A. Rashid, L. Dakuna, H. Louie, D. Ng, P. Vandamme, W. Johnson and V. L. Chan, J. Clin. Microbiol., 2000, 38, 1488–1494 CAS.
- M. Lund, S. Nordentoft, K. Pedersen and M. Madsen, J. Clin. Microbiol., 2004, 42, 5125–5132 CrossRef CAS.
- J. D. Klena, C. T. Parker, K. Knibb, J. C. Ibbitt, P. M. L. Devane, S. T. Horn, W. G. Miller and M. E. Konkel, J. Clin. Microbiol., 2004, 42, 5549–5557 CrossRef CAS.
- G. Wang, C. G. Clark, T. M. Taylor, C. Pucknell, C. Barton, L. Price, D. L. Woodward and F. G. Rodgers, J. Clin. Microbiol., 2002, 40, 4744–4747 CrossRef CAS.
- K. E. Dingle, F. M. Colles, D. R. A. Wareing, R. Ure, A. J. Fox, F. E. Bolton, H. J. Bootsma, R. J. L. Willems, R. Urwin and M. C. J. Maiden, J. Clin. Microbiol., 2001, 39, 14–23 CrossRef CAS.
- D. S. Andrew, S. Bala and I. F. Patricia, J. Clin. Microbiol., 2003, 41, 4733–4739 CrossRef.
- R. S. Y. Wong and A. W. Chow, FEMS Microbiol. Lett., 2002, 206, 107–113 CAS.
- A. D. Sails, F. J. Bolton, A. J. Fox, D. R. A. Wareing and D. L. A. Greenway, Mol. Cell. Probes, 1998, 12, 317–322 CrossRef CAS.
- S. L. On and P. J. Jordan, J. Clin. Microbiol., 2003, 41, 330–336 CrossRef CAS.
- P. Vandamme, in Campylobacter, ed. I. Nachamkin and M. J. Blaser, American Society for Microbiology, Washington, DC, 2000, pp. 3–26 Search PubMed.
- J. A. Frost, J. Appl. Microbiol., 2001, 90, 85S–95S CrossRef.
- M. Schena, D. Shalon, R. Davis and P. O. Brown, Science, 1995, 270, 467–470 CrossRef CAS.
- P. O. Brown and D. Botstein, Nat. Genet., 1999, 21, 33–37 CrossRef CAS.
- D. J. Duggan, M. Bittner, Y. Chen, P. Meltzer and J. M. Trent, Nat. Genet., 1999, 21, 10–14 CrossRef CAS.
- T. R. Golub, D. K. Slonim, P. Tamayo, C. Huard, M. Gaasenbeek, J. P. Mesirov, H. Coller, M. L. Loh, J. R. Downing, M. A. Caligiuri, C. D. Bloomfield and E. S. Lander, Science, 1999, 286, 531–537 CrossRef CAS.
- A. A. Alizadeh, M. B. Eisen, R. E. Davis, C. Ma, I. S. Lossos, A. Rosenwald, J. C. Boldrick, H. Sabet, T. Tran, X. Yu, J. I. Powell, L. Yang, G. E. Marti, T. Moore, J. Hudson, Jr., L. Lu, D. B. Lewis, R. Tibshirani, G. Sherlock, W. C. Chan, T. C. Greiner, D. D. Weisenburger, J. O. Armitage, R. Warnke, R. Levy, W. Wilson, M. R. Grever, J. C. Byrd, D. Botstein, P. O. Brown and L. M. Staudt, Nature, 2000, 403, 503–511 CrossRef CAS.
- K. Lindroos, S. Sigurdsson, K. Johansson, L. Ronnblom and A.-C. Syvanen, Nucleic Acids Res., 2002, 30, e70 CrossRef.
- M. Erali, B. Schmidt, E. Lyon and C. Wittwer, Clin. Chem., 2003, 49, 732–739 CrossRef CAS.
- E. M. Weidenhammer, B. F. Kahl, L. Wang, L. Wang, M. Duhon, J. A. Jackson, M. Slater and X. Xu, Clin. Chem., 2002, 48, 1873–1882 CAS.
- N. Nagan and D. J. O'Kane, Clin. Biochem., 2001, 34, 589–592 CrossRef CAS.
- E. A. Barlaan, M. Sugimori, S. Furukawa and K. Takeuchi, J. Biotechnol., 2005, 115, 11–21 CrossRef CAS.
- K. Zimmermann, T. Eiter and F. Scheiflinger, J. Microbiol. Methods, 2003, 55, 471–474 CrossRef CAS.
- Y. Huang, J. Shirajian, A. Schroder, Z. Yao, T. Summers, D. Hodko and R. Sosnowski, Electrophoresis, 2004, 25, 3106–3116 CrossRef CAS.
- K. L. F. Cooper and R. V. Goering, J. Mol. Diagn., 2003, 5, 28–33 Search PubMed.
- B. C. Kim, J. H. Park and M. B. Gu, Anal. Chem., 2005, 77, 2311–2317 CrossRef CAS.
- B. C. Kim, J. H. Park and M. B. Gu, Environ. Sci. Technol., 2004, 38, 6767–6774 CrossRef CAS.
- J. R. E. Shepard, Y. Danin-Poleg, Y. Kashi and D. R. Walt, Anal. Chem., 2005, 77, 319–326 CrossRef CAS.
- S. Ahn and D. R. Walt, Anal. Chem., 2005, 77, 5041–5047 CrossRef CAS.
- L. Wu, D. K. Thompson, X. Liu, M. W. Fields, J. M. Tiede and J. Zhou, Environ. Sci. Technol., 2004, 38, 6775–6782 CrossRef CAS.
- N. Sergeev, M. Distler, S. Courtney, S. F. Al-khaldi, D. Volokhov, V. Chizhikov and A. Rasooly, Biosens. Bioelectron., 2004, 20, 684–698 CrossRef CAS.
- G. Keramas, D. D. Bang, M. Lund, M. Madsen, H. Bunkenborg, P. Telleman and C. B. V. Christensen, J. Clin. Microbiol., 2004, 42, 3985–3991 CrossRef CAS.
- G. E. C. Sheridan, C. I. Masters, J. A. Shallcross and B. M. Mackey, Appl. Environ. Microbiol., 1998, 64, 1313–1318 CAS.
- G. M. Ransom and B. E. Rose, in USDA/FSIS Microbiology Laboratory Guidebook, ed. B. P. Dey and C. P. Lattuada, USDA/FSIS, Department of Agriculture, Food Safety and Inspection Services, Office of Public Health and Science, Microbiology Division, Washington, DC, USA, 3rd edn, 1998, ch. 6, pp. 6-1–6-10. Available online at http://www.fsis.usda.gov/Ophs/Microlab/Mlgchp6.pdf, retrieved on 22/02/2006 Search PubMed.
- M. A. VanderKop, MSc thesis, University of Alberta, 2003.
- J. W. Messer, T. F. Midura and J. T. Peeler, in Compendium of Methods for the Microbiological Examination of Foods, American Public Health Association, Washington, DC, 1992, pp. 25–49 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |