Single molecule studies of DNA binding proteins using optical tweezers
Yuji
Kimura
ab and
Piero R.
Bianco
*abc
aCenter for Single Molecule Biophysics, University at Buffalo, Buffalo, NY 14214, USA
bDepartment of Microbiology and Immunology, University at Buffalo, Buffalo, NY 14214, USA
cDepartment of Biochemistry, University at Buffalo, Buffalo, NY 14214, USA. E-mail: pbianco@buffalo.edu; Fax: (716)829-2158; Tel: (716)829-2599
First published on 30th June 2006
Abstract
Optical tweezers have become a versatile tool in the biological sciences. Combined with various types of optical microscopy, they are being successfully used to discover the fundamental mechanism of biological processes. Recently, the study of proteins acting on DNA was aggressively undertaken at the single-molecule level. Here, we review the most recent studies which have revealed the dynamic behavior of individual protein molecules at work on DNA, providing detailed mechanistic insight that could not be revealed, at least not easily, using bulk-phase or ensemble approaches.
Introduction
Since optical tweezers were invented by Arthur Ashkin,1,2 they have found increasing application in biological studies. Optical tweezers can trap objects in the nanometre to micrometre size range, and manipulate trapped objects with sub-nanometre accuracy3 although nanometre resolution is more typical. More importantly, optical tweezers are compatible with various types of light microscopy, such as bright field, differential interference contrast, phase contrast and fluorescence.4 These features have allowed optical tweezers to become one of the most successful single-molecule techniques used in biological science.Initially, optical tweezers were applied to the single-molecule investigation of cytoskeletal motor proteins.5,6 Recent advances have made it possible to study DNA binding proteins at the single-protein level. This important group of proteins includes those which affect conformational changes in nucleic acids, as well as energy-fueled molecular motors. In this review, the principle of the optical trap is explained briefly. Then, the application of optical tweezers to the study of DNA binding proteins is presented.
Principle of optical tweezers
Typically, optical traps or optical tweezers are formed by focusing a Gaussian-profiled infrared laser beam (λ = 1064 nm) through a high numerical aperture, microscope objective to a diffraction-limited spot in the specimen plane of a microscope. As a result, a steep, three-dimensional gradient in light intensity forms near the focal point that has the ability to optically trap small particles.The ability to trap and manipulate small objects, such as polystyrene beads, results from light possessing momentum which is in the direction of propagation of the beam (Fig. 1). When the direction of light is altered by a particle via reflection or refraction, a corresponding change in momentum occurs. The law of conservation of momentum requires that the bead must undergo an equal and opposite momentum change, giving rise to forces acting on the particle.
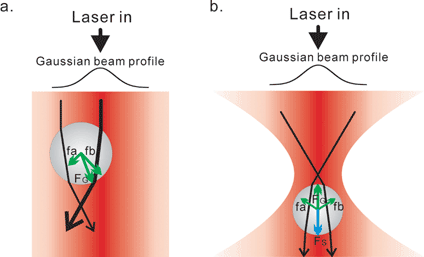 | ||
| Fig. 1 Schematics showing the principle of optical tweezers based on ray optics. The ability to trap and manipulate small objects, such as polystyrene beads, results from light possessing momentum which is in the direction of propagation of the beam. Here, a bead is illuminated by a Gaussian profiled laser beam. The representative laser paths are shown as black lines with arrows indicating the direction of beam propagation. The thickness of the black lines indicates the intensity of laser beam. The forces are shown as green and blue lines with arrows indicating the direction of forces. The length of the lines indicates the intensity of forces. (a) Gradient forces which are generated upon refraction (FG, fa and fb, green lines). The beam is refracted on the surface of the bead, resulting in the change of momentum of the beam. Gradient forces (FG) result to compensate the momentum changes on the surface of the bead. The gradient forces from the inner region (fb) are larger than that from outer region (fa) of the beam, due to the profile of the laser. Consequently, the net gradient force in the lateral direction directs particles to center of the beam (FG). (b) Stable 3D trapping. The laser beam was focused by a lens of high numerical aperture. The scattering forces (Fs, blue line) are in the direction of propagation of the laser beam (i.e., downward in this figure), while the gradient forces are directed toward the focused spot. Consequently, the bead is trapped slightly beyond the focused spot where the gradient force and scattering force are in equilibrium. | ||
Small particles (ranging in size from 10 nm to 10 µm in diameter) experience two types of forces near the focus which results in their stable three-dimensional trapping. Scattering forces arise from reflection of light at the surface of the particle, pushing the particles along the path of the laser beam in the direction of propagation of the light. In opposition to these, gradient forces tend to draw particles towards the center of the trap, thereby preventing their escape. Once the gradient forces dominate, a stable three-dimensional trap results. Schematics showing these forces are presented in Fig. 1 and the reader is referred to ref. 4 for a more detailed description.
The ability to trap in three dimensions has been taken advantage of in biological single-molecule studies. Here, trapped particles are often micron-sized polystyrene beads to which protein and/or DNA molecules are attached via non-covalent bonds such as those between avidin and biotin or between an antigen and an antibody. In addition to being combined with different types of optical microscopy, optical tweezers can be set up in a variety of configurations with each being unique: (1) a single optical trap set up/coverslip pioneered by the Block lab for kinesin6 and advanced by the same lab for RNA polymerase7 and the Wang lab for nucleosome studies;8–10 (2) a dual trap configuration with molecules suspended between them, pioneered by the Spudich lab for myosin5 and adapted to DNA motors by the Block lab;3,11 (3) a single trap combined with a suction pipette pioneered by the Bustamante group12 for use in DNA and RNA dynamics and adapted for DNA motors; (4) a single optical trap combined with a laminar flow developed originally by the Chu lab for the analysis of DNA polymer dynamics13 and (5) a single trap positioned in a multi-channel laminar flow cell pioneered by the Kowalczykowski lab for the study of DNA helicases.14–16 Configurations 1–3 have been used predominantly for force measurements while configurations 4 and 5 have been used for visualization of DNA and protein-mediated dynamics of DNA using fluorescence video-microscopy. Using these configurations, optical tweezers have permitted detailed analysis of a variety of proteins as described below.
Application to biological motors
The first biological motors studied at the single-molecule level with optical tweezers were cytoskeletal motor proteins.5,6 These motor proteins (i.e., myosin, kinesin and dynein) generate mechanical force to move along protein tracks (for example, actin filaments and microtubules), using energy liberated from the hydrolysis of ATP. Force generation allows these motor proteins to engage in work inside cells, including that required to perform chromosome segregation, vesicle transport and muscle contraction.17 Using optical tweezers, precise measurement of the fundamental step size and maximum force generated by single molecules of these motor protein molecules has been achieved (for example, the step size and maximum force are 8 nm and 6 pN for kinesin, respectively).18,19For these studies, the optical tweezers were used to manipulate individual motor protein molecules via direct attachment to micrometre-sized polystyrene beads. The purpose of the beads is two-fold. First, they are used to deliver the motor to its tracks. Second, they are used to monitor the position of the bead-attached motor proteins most recently with an accuracy of ∼0.1 nm, far beyond the diffraction limit of light.3 To track bead movement, the bright-field image of the bead is projected onto a photodetector, such as a quadrant photodiode or position sensitive detector, and the change in position of the image is measured (i.e., nanometry).4 Alternatively, the interference pattern of the bead can be projected onto a photodetector, where the change in the pattern is converted to a position signal to monitor bead movement (i.e., interferometry).20 Furthermore, and simultaneously, the measured displacement in each case can be converted to force exerted by the motor protein.4 As an application, a sensitive feedback system is being used to measure the position of a molecular motor under a constant load. When the bead attached to the molecular motor pulls with a force greater than a preset level, the position of the optical trap is moved to decrease the force. The error signal generated gives the position of the molecular motor.20
Force measurement is made possible due to the spring-like nature of the optical trap. As described above and shown in Fig. 1, a trapped bead is pulled back toward the trap center when displaced from the trap center by an external force. The force exerted on the bead varies linearly with its displacement from trap center over a limited range of approximately 200 nm for a 1 µm diameter bead, giving a spring constant. From the spring constant, the force generated by the individual motor protein can be calculated. Thus, the measurement of displacement and force is done simultaneously. As for cytoskeletal motors, optical tweezers have made it possible to observe the detailed behavior of DNA binding proteins as described below.
Nucleic-acid polymerases
This family of DNA motor proteins plays essential roles in the life of an organism. While translocating along a DNA template (that is, a DNA track), DNA polymerases faithfully replicate DNA while RNA polymerases faithfully transcribe DNA into RNA.RNA polymerase (RNAP) is a highly processive motor, translocating thousands of base pairs without detaching from the DNA template. The energy used to drive RNAP translocation comes from the nucleoside triphosphate addition to the 3′-end of the nascent RNA molecule.21 During translocation, RNAP must accurately read the sequence of the template strand and transcribe mRNA that is subsequently translated into protein. Transcription occurs with high fidelity, and by comparison to DNA polymerase, must include a proofreading mechanism, as suggested by bulk-phase studies.22 During proofreading, the enzyme would be expected to transiently pause and possibly even backtrack to allow removal of misincorporated bases.
To provide detailed insight into transcription fidelity, a series of studies were done using initially, the single-trap coverslip configuration7 and subsequently, the dual trap configuration to minimize noise3,11 (Fig. 2). The results revealed that the translocation velocity varied significantly between individual RNAP molecules.7,11 Second, for individual polymerase molecules, translocation was non-uniform, as traces of individual enzymes exhibited periods of constant velocity interspersed with several pauses of various duration. The pauses were classified based on their duration. Longer pauses had lifetimes greater than 20 seconds.11 They were distributed uniformly, occurring on average once every 1000 bp transcribed. Importantly, individual RNAP molecules were demonstrated to be capable of back tracking a distance of approximately 5 bp consistent with anticipated proofreading. In order to test whether backtracking was coupled to proofreading, GreA and GreB were added to the reaction. These proteins are transcription elongation factors which induce cleavage of nascent RNA.23 After backtracking, by cleaving the nascent RNA still bound to RNAP, the end of the RNA where transcription should be reinitiated is repositioned closer to the active site. This would be expected to enhance the rate of reinitiation and be observed as an overall decrease in pause duration. Consequently, in the presence of GreA and GreB, both the frequency and the duration of pauses were found to decrease. Therefore, the observed backtracking is consistent with a proofreading mechanism, which consists of rearward movement followed by nucleolysis of RNA.
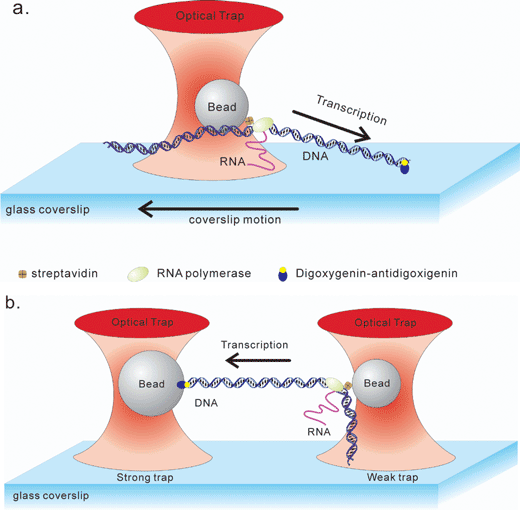 | ||
| Fig. 2 Optical tweezers configurations used to study RNA polymerase. (a) A single trap configuration is shown. One end of the DNA is attached to a glass coverslip via a digoxigenin–antidigoxigenin linkage, while RNA polymerase is attached to an optically trapped bead via an avidin–biotin bond.7 The bead position is maintained by stage motion to provide constant tension (typically from right to left). (b) A schematic of a dual-trap configuration is shown.3,11 An RNA polymerase–DNA complex is trapped by two optical traps simultaneously. The left DNA end is manipulated via a digoxigenin–antidigoxigenin linkage, while RNA polymerase is attached via an avidin–biotin interaction. In this figure, the upstream end of DNA is linked to the left bead so that RNAP transcribes from right to left. The tension of the DNA–RNAP complex was kept constant during transcription by moving left stronger trap. Figures are adapted from ref. 3, 7 and 11. | ||
The second class of pauses was short-lived, with a duration of 25 seconds or less, occurring with a frequency of ∼10 events every 1000 bp. These constituted > 95% of the pauses observed,7 with duration and frequency unaffected by load applied to the enzyme by the optical tweezers, from −37 to +27 pN (negative and positive values indicates hindering and assisting force for translocation, respectively). This is in contrast to the long-lived pauses described above, where frequency decreased from 1 event to 0.03 events every 1000 bp under load of +8 pN. The independence of the duration from load showed that motion accompanying the short pauses was as small as 0.06 bp, hence the short-lived pauses do not correspond to backtracking motion. Instead, the short-lived pause is a transient state, which could potentially precede long-lived pauses.
Most recently, the individual steps taken by RNAP during transcription were observed.3 The study demonstrated that RNAP advances predominantly in 3.7 Å (0.37 nm) increments along DNA. The distance of 3.7 Å is similar to that of one base pair in B-form DNA (i.e., 3.4 Å), suggesting that RNAP advances one base pair each time a nucleotide is incorporated. Thus, during transcription, RNA polymerase translocates in increments of predominantly 1 base pair. When incorrect bases are incorporated, it pauses and backtracks to facilitate base removal, thereby ensuring transcription occur with high fidelity.
DNA helicases
DNA helicases are ubiquitous enzymes that function to unwind DNA duplexes into their component single strands in a reaction that is coupled to the hydrolysis of nucleoside 5′-triphosphates.24,25 DNA unwinding serves to provide nascent, single-stranded DNA for the processes of DNA repair, replication and recombination.One of the most well studied DNA helicases is the RecBCD enzyme of E. coli, a heterotrimeric enzyme that has both helicase and nuclease activities.26 RecBCD is composed of 3 subunits, RecB, RecC and RecD. RecB and RecD are motor subunits that drive translocation of RecBCD along double-stranded DNA (dsDNA, Fig. 3a). RecC is responsible for strand separation and recognition of an octanucleotide sequence known as chi (chi = crossover hotspot instigator, 5′-GCTGGTGG-3′ or χ). Previous work demonstrated that chi is responsible for regulating the nuclease activity of RecBCD.26 Further, the interaction with chi was proposed to result in the ejection of the RecD subunit,27 although this was controversial.28
 | ||
| Fig. 3 RecBCD translocation assay. (a) A schematic showing the regulation of the nuclease activity of the translocating RecBCD enzyme by the recombination hotspot χ. Translocation and unwinding initiates at dsDNA ends. As RecBCD translocates and unwinds the DNA, the 3′-terminated strand relative to the entry point of the enzyme (the top strand as shown) is degraded more vigorously than the 5′-terminated strand. When RecBCD recognizes χ, the nuclease activity on the 3′-terminated strand is attenuated, while that on the 5′-terminated strand is upregulated. Continued translocation by RecBCD results in the formation of a DNA molecule with a 3′ single-stranded tail with the χ-sequence positioned close to the 3′-terminus of the tail. (b) A schematic showing the arrangement of the laminar flow cell and syringe pump used to visualize individual RecBCD enzymes14–16 (adapted from ref. 14). A syringe pump introduces solutions into a two channel flow cell to create conditions of laminar flow where the 2 solutions flow parallel to one another with negligible mixing, as indicated by the dashed line. The inset shows a magnified view of the oval area in the flow cell; the position of the optical trap is designated by X. After trapping a single RecBCD–DNA–bead complex (inset), the stage is moved (black arrow) thereby translating the complex (blue arrow) to the reaction side to initiate translocation and DNA unwinding. As unwinding and degradation of DNA proceed, fluorescent dye molecules (YOYO-1, light green stars) are displaced, resulting in loss of fluorescence (black stars). Translocation and DNA unwinding by individual RecBCD molecules is detected as a change in a length of the fluorescent DNA molecule. The translocation of RecBCD in ref. 15 was monitored directly by tracking the position of a fluorescent bead attached to the RecD subunit. The experiments in ref. 14 were done in the absence of chi (χ0) and those for ref. 15 and 16 used DNA containing chi (χ+). The small arrow above χ designates the orientation from which RecBCD must approach χ in order to recognize and be regulated by this sequence. | ||
Translocation and unwinding by RecBCD initiates uniquely at dsDNA ends, with the RecB and RecD subunits interacting with the 3′- and 5′-terminated strands of dsDNA, respectively (Fig. 3a). RecB and RecD translocate with opposite polarity and thereby drive the motion of the RecBCD holoenzyme in one direction along dsDNA, due to the anti-parallel nature of duplex DNA strands. Translocation is accompanied by the unwinding and degradation of DNA, with the 3′-terminated strand relative to the entry site of the enzyme being degraded more vigorously than 5′-terminated strand (Fig. 3a). Upon χ-recognition, the nuclease activity on the 3′-terminated strand is attenuated, while that on the 5′-terminated strand is upregulated. Continued translocation past χ results in the formation of DNA with a 3′ single-stranded tail with the chi sequence positioned close to the 3′-terminus of the tail. This tailed DNA molecule is a key, early recombination intermediate onto which the RecA protein is loaded.
To elucidate the mechanism of action of this complex enzyme and its regulation by χ, a series of single-molecule investigations were undertaken.14–16 Using a combination of optical tweezers and two-channel, micro-machined laminar flow cells, dsDNA translocation and unwinding;14 χ-recognition;15 and the response of RecBCD to χ15,16 were examined at the single enzyme-DNA level (Fig. 3b). Here, optical tweezers were used to isolate individual enzyme DNA–bead complexes. Isolation and manipulation was facilitated via 1 µm streptavidin-coated polystyrene beads attached to one end of asymmetrically biotinylated DNA. Under these conditions, RecBCD binds to the free end of dsDNA distal to the bead, forming an initiation complex. These complexes were introduced into the first channel of a flow cell, with the dsDNA extended by laminar flow and visualized using the intercalating fluorophore, YOYO-1 (Fig. 3b). Reaction buffer containing ATP was introduced into the second channel under conditions of laminar flow which creates streams that flow parallel to one another with negligible mixing. By translocating the optically trapped bead–DNA–RecBCD complex into the second channel beyond the boundary between the two flows, translocation and unwinding by RecBCD was initiated, visualized as a decrease in the length of the dsDNA14 (Fig. 3b, inset).
In the absence of χ, translocation and unwinding by individual RecBCD molecules was continuous and exhibited no detectable pausing.14 Similar to RNA polymerase, the rate of unwinding by individual enzymes varied by as much as five-fold. In contrast, the encounter with χ results in not only an alteration in nuclease activity (demonstrated thus far only in bulk-phase assays), but also in a pause of the translocating enzyme.16 The duration of the pause was as long as 5.4 s on average, when the chi sequence was 8229 bp from the DNA end; pausing had not been detected so far using bulk phase biochemical assays. Once the enzyme re-established translocation, surprisingly, the rate decreased by approximately 50% from 598 to 309 bp s−1 (pre- and post chi-recognition rate, respectively).16 The change in translocation rate resulting from χ-recognition could be due to the ejection of RecD subunit. To address this possibility, RecD was specifically and uniquely labeled by a fluorescent bead via a biotin–avidin interaction. Then, the presence of the RecD subunit in the translocating enzyme was assessed before and after χ recognition. The results show that the decreased translocation rate was not due to the loss of the RecD subunit as RecD was observed to cotranslocate with RecBC after recognition of χ.15 Instead, the change in translocation rate is due to a χ-induced conformational change within the RecBCD complex.15 The nature of the conformational change is yet to be elucidated. Thus these studies revealed that a rapidly translocating DNA motor can recognize and respond to an octanucleotide sequence, resulting in substantial changes in the enzyme. These changes in RecBCD facilitate the accurate repair of dsDNA breaks via homologous recombination.26
Nucleosomes
In the eukaryotic nucleus, genomic DNA is packaged into a highly compacted DNA–protein complex called chromatin (see ref. 29 and ref. therein). The basic repeating unit of chromatin is the nucleosome, which contains 146 bp of DNA wrapped around an octamer of histone proteins in 1.65 turns of a left-handed superhelix. By wrapping the DNA, nucleosomes package the genome into a manageable space inside the nucleus. Nucleosomes are further arranged into a more complex hierarchical structure. In addition to their condensation function, nucleosomes play an important role in all aspects of DNA metabolism. By disassembling or repositioning of nucleosomes, accessibility of the previously compacted DNA is altered, thereby providing access to DNA based enzymes.Due to the complex structure of chromatin and its dynamic nature, a study of the behavior of individual nucleosomes within chromatin is highly challenging using conventional techniques. To study nucleosome behavior, a single chromatin fiber was used, in which the response of nucleosomal DNA to applied external tension was studied, initially using single optical trap in combination with a suction pipette.30,31 Recent studies were undertaken using a single optical trap set-up (Fig. 4a).8–10 Here, one end of an individual chromatin fiber (i.e., an array of nucleosomes on a single DNA molecule) was fixed to the surface of a coverslip while the opposite end was manipulated by the optical trap via a polystyrene bead. By moving the coverslip relative to the position of the trap, the chromatin fiber was stretched and tension was applied. As the distance between the trap and the site of attachment increased, the applied tension increased accordingly, resulting in release of DNA from the histone core proteins. DNA release occurred in 2 stages correlating with low and high affinity modes of binding to the histone core (Fig. 4b). The low affinity mode was observed during the initial release phase at low external force (i.e., <15 pN). DNA release occurred gradually as the applied force disrupted the uniformly-distributed interactions between outer turns of DNA and histone proteins within the nucleosomes. At higher force (i.e., >15 pN), the remaining DNA was released abruptly with peak forces, correlating with the higher affinity DNA binding mode between inner turns of DNA and histone proteins. Subsequent work demonstrated that acetylation of the histone core proteins decreased both the amount of DNA bound to the histone core by 23% and the amount of peak force required to remove the DNA completely by 1.8 pN.8 These results indicate that histone acetylation controls accessibility of DNA to enzymes by reducing the affinity of histone proteins for the DNA.
 | ||
| Fig. 4 Nucleosome dynamics revealed by disruption of individual nucleosomes. (a) A schematic showing experimental configuration used for this study (adapted from ref. 10). Similar to the RNA polymerase study, one end of a chromatin fiber was fixed to a glass coverslip via a digoxigenin–antidigoxigenin linkage, while the other end was attached to an optically trapped bead via an avidin–biotin bond.8–10 To apply tension, the chromatin fiber was stretched by moving the coverslip relative to the position of the trap. (b) Force–extension curve of a single chromatin fiber (reprinted from ref. 10, Copyright 2002 National Academy of Sciences, USA). The coverslip was moved with constant velocity. DNA release started gradually in the low force range. Then, in the higher force range (>15 pN), the remaining DNA release occurred abruptly, giving a saw tooth pattern composed of 17 disruption peaks, which indicated 17 disruptions of nucleosome core particles. The force–extension curve of a naked DNA molecule had no disruption peak (red dotted line). | ||
Summary
Optical tweezers have contributed to the discovery of the dynamic behavior of individual protein molecules at the single-molecule level. DNA binding proteins are no exception, with studies providing an unprecedented level of detailed insight. Moreover, the number of DNA binding proteins being studied at this level is continuing to grow with a dsDNA translocase (FtsK from E. coli) and a bacteriophage DNA packaging motor being added to the list.32,33 In the coming years, further modifications to optical tweezer methods, capable of controlling the torque in the DNA34,35 or applying constant force using optical methods instead of a mechanical feed-back system,36,37 will also be applied to single-molecule studies of DNA binding proteins. Due to their broad adaptability, optical tweezers will undoubtedly continue to contribute significant insight to the understanding of protein–nucleic acid interactions, including those of ribosomes, the mRNA splicing process, DNA mismatch repair and recombination reactions.Acknowledgements
We thank Cuiling Xu and Jackson Buss for critical reading of the manuscript. The work in the laboratory of P.R.B. was supported by NIH grant GM66831-01.References
- A. Ashkin and J. M. Dziedzic, Science, 1987, 235, 1517–1520 CrossRef CAS.
- A. Ashkin, J. M. Dziedzic, J. E. Bjorkholm and S. Chu, Opt. Lett., 1986, 11, 288–290 Search PubMed.
- E. A. Abbondanzieri, W. J. Greenleaf, J. W. Shaevitz, R. Landick and S. M. Block, Nature, 2005, 438, 460–465 CrossRef CAS.
- M. P. Sheetz, Laser Tweezers in Cell Biology, Academic Press, London, UK, 1998 Search PubMed.
- J. T. Finer, R. M. Simmons and J. A. Spudich, Nature, 1994, 368, 113–119 CrossRef CAS.
- K. Svoboda, C. F. Schmidt, B. J. Schnapp and S. M. Block, Nature, 1993, 365, 721–727 CrossRef CAS.
- K. C. Neuman, E. A. Abbondanzieri, R. Landick, J. Gelles and S. M. Block, Cell, 2003, 115, 437–447 CrossRef CAS.
- B. Brower-Toland, D. A. Wacker, R. M. Fulbright, J. T. Lis, W. L. Kraus and M. D. Wang, J. Mol. Biol., 2005, 346, 135–146 CrossRef CAS.
- B. Brower-Toland and M. D. Wang, Methods Enzymol., 2004, 376, 62–72 CAS.
- B. D. Brower-Toland, C. L. Smith, R. C. Yeh, J. T. Lis, C. L. Peterson and M. D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1960–1965 CrossRef CAS.
- J. W. Shaevitz, E. A. Abbondanzieri, R. Landick and S. M. Block, Nature, 2003, 426, 684–687 CrossRef CAS.
- C. Bustamante, Z. Bryant and S. B. Smith, Nature, 2003, 421, 423–427 CrossRef.
- T. T. Perkins, S. R. Quake, D. E. Smith and S. Chu, Science, 1994, 264, 822–826 CrossRef CAS.
- P. R. Bianco, L. R. Brewer, M. Corzett, R. Balhorn, Y. Yeh, S. C. Kowalczykowski and R. J. Baskin, Nature, 2001, 409, 374–378 CrossRef CAS.
- N. Handa, P. R. Bianco, R. J. Baskin and S. C. Kowalczykowski, Mol. Cell, 2005, 17, 745–750 CrossRef CAS.
- M. Spies, P. R. Bianco, M. S. Dillingham, N. Handa, R. J. Baskin and S. C. Kowalczykowski, Cell, 2003, 114, 647–654 CrossRef CAS.
- J. Howard, Mechanics of Motor Proteins and the Cytoskeleton, Sinauer Associates, Inc., Sunderland, MA, USA, 2001 Search PubMed.
- Y. Ishii and T. Yanagida, Cell. Mol. Life Sci., 2002, 59, 1767–1770 CrossRef CAS.
- A. Yildiz and P. R. Selvin, Trends Cell Biol., 2005, 15, 112–120 CrossRef CAS.
- K. C. Neuman and S. M. Block, Rev. Sci. Instrum., 2004, 75, 2787–2809 CrossRef CAS.
- R. A. Mooney, I. Artsimovitch and R. Landick, J. Bacteriol., 1998, 180, 3265–3275 CAS.
- E. Nudler, J. Mol. Biol., 1999, 288, 1–12 CrossRef CAS.
- S. Borukhov, V. Sagitov and A. Goldfarb, Cell, 1993, 72, 459–466 CrossRef CAS.
- E. Delagoutte and P. H. von Hippel, Q. Rev. Biophys., 2002, 35, 431–478 CrossRef CAS.
- E. Delagoutte and P. H. von Hippel, Q. Rev. Biophys., 2003, 36, 1–69 CrossRef CAS.
- D. A. Arnold and S. C. Kowalczykowski, RecBCD Helicase/Nuclease, in Encyclopedia of Life Sciences, John Wiley & Sons, Ltd, Chichester, 1999, http://www.els.net/, DOI:10.1038/npg.els.0000586.
- F. W. Stahl, L. C. Thomason, I. Siddiqi and M. M. Stahl, Genetics, 1990, 126, 519–533 Search PubMed.
- D. G. Anderson, J. J. Churchill and S. C. Kowalczykowski, Genes Cells, 1997, 2, 117–128 CrossRef CAS.
- A. Lusser and J. T. Kadonaga, Nat. Methods, 2004, 1, 19–26 CrossRef CAS.
- M. L. Bennink, S. H. Leuba, G. H. Leno, J. Zlatanova, B. G. de Grooth and J. Greve, Nat. Struct. Biol., 2001, 8, 606–610 CrossRef CAS.
- Y. Cui and C. Bustamante, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 127–132 CrossRef CAS.
- Y. R. Chemla, K. Aathavan, J. Michaelis, S. Grimes, P. J. Jardine, D. L. Anderson and C. Bustamante, Cell, 2005, 122, 683–692 CrossRef CAS.
- P. J. Pease, O. Levy, G. J. Cost, J. Gore, J. L. Ptacin, D. Sherratt, C. Bustamante and N. R. Cozzarelli, Science, 2005, 307, 586–590 CrossRef CAS.
- C. Claudet and J. Bednar, Appl. Opt., 2005, 44, 3454–3457 CrossRef.
- A. La Porta and M. D. Wang, Phys. Rev. Lett., 2004, 92, 190801 CrossRef.
- W. J. Greenleaf, M. T. Woodside, E. A. Abbondanzieri and S. M. Block, Phys. Rev. Lett., 2005, 95, 208102 CrossRef.
- R. Nambiar, A. Gajraj and J. C. Meiners, Biophys. J., 2004, 87, 1972–1980 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |